






前言
研究背景
心肌梗死是全球致死致残的主要心血管疾病,即便实施再灌注治疗,患者仍易出现不良心室重构与心力衰竭,其核心原因在于心肌损伤后修复机制不足。巨噬细胞介导的胞葬作用是清除凋亡心肌细胞、驱动修复型巨噬细胞转化、促进炎症消退的关键环节,高效胞葬可显著改善心梗后组织修复,但调控该过程的关键分子机制尚不明确。CD40作为肿瘤坏死因子受体超家族成员,已知通过TRAF2/3/5 与TRAF6 两个位点传递信号,其中TRAF6介导促炎反应,而CD40-TRAF2/3/5在巨噬细胞中的功能、以及两条通路在心梗后胞葬与修复中的作用仍未被阐明。本研究旨在揭示CD40 及其下游信号对心梗后巨噬细胞胞葬的调控机制,明确关键通路与效应分子,为寻找心肌缺血损伤的治疗靶点、开发精准免疫调控策略提供重要理论依据。

发表期刊:Circulation
影响因子:38.6
涉及的欧易生物服务产品:单细胞转录组测序
技术路线

关键发现与实验结论
Result1 心梗后CD40高表达且主要来源于髓系巨噬细胞
心肌梗死后,CD40主要由梗死区髓源性巨噬细胞特异性高表达,且在小鼠与人的心肌梗死模型中呈现一致的表达规律。
野生型 C57BL/6 小鼠心肌梗死模型中,CD40在心梗后3–7天的梗死区组织中表达显著上调并达峰,随后逐渐回落;蛋白与流式检测证实,该上调主要发生于髓源性巨噬细胞(Fig 1A–D)。免疫荧光共染进一步明确,CD40主要定位于梗死区的CCR2⁺髓源性巨噬细胞(Fig 1E–G)。
人心肌梗死组织的免疫组化染色与公共数据库 scRNA-seq 分析结果与小鼠模型结果高度一致(Fig 1I–L)。

图1 小鼠心梗模型与缺血性心肌病患者心脏巨噬细胞 CD40 表达升高
Result2 CD40 缺失加重心梗后的心脏损伤并损害心功能与修复过程
CD40缺失会显著恶化心梗后心功能、扩大梗死面积并加重不良心室重构,提示巨噬细胞表达的CD40是心梗后心脏修复的关键保护分子。
心脏超声与MRI检测显示,CD40全身敲除小鼠心梗后28天的左室射血分数(LVEF)、短轴缩短率(FS)均显著低于野生型小鼠,心室扩张与收缩功能障碍更为严重(Fig 2A–E)。
TTC 与 Masson染色证实CD40 敲除小鼠心梗面积与纤维化区域显著增大(Figure 2F、2G)。

图2 全身与髓系特异性敲除 CD40 会扩大心梗面积并损害心梗后心功能
Result3 CD40缺失损害凋亡心肌细胞的清除
CD40 缺失会显著抑制心梗后巨噬细胞的胞葬作用,导致凋亡心肌细胞清除障碍,直接阻断修复型巨噬细胞的极化进程。
小鼠心梗后心脏巨噬细胞的单细胞测序(scRNA-seq)显示,CD40 缺失导致具有胞葬与修复功能的巨噬细胞前体亚群数量锐减,促炎亚群比例相对升高(Fig 3A-B)
心梗后第 3 天的TUNEL染色显示,CD40 敲除小鼠梗死区凋亡心肌细胞数量显著多于野生型小鼠,提示凋亡细胞清除受阻(Fig 3D-F)。
体外胞葬实验与流式分析证实,CD40 缺失的髓源巨噬细胞(BMDM)吞噬凋亡心肌细胞的能力显著下降,同时胞葬关键分子 MerTK、Arg1的表达水平明显降低(Fig 3J-N)。

图3 全身与髓系特异性敲除 CD40 会损害心梗后凋亡心肌细胞的清除
Result4 CD40缺失损害心肌梗死后修复性巨噬细胞极化
CD40缺失通过减少修复表型巨噬细胞前体数量,阻断其向修复表型巨噬细胞的分化,从而延迟炎症消退并损害心脏修复。
scRNA-seq分析显示CD40 KO后cluster0细胞显著减少,其标记基因与胞葬通路高度相关(Figure 4G、4H)。伪时间分析证实cluster 0为修复性巨噬细胞(Mac 1)的直接前体,CD40缺失导致该分化路径受阻(Figure 4I–4O)。
流式细胞术显示CD40 KO和CD40 cko小鼠心脏中CD206+修复巨噬细胞比例显著降低,Ly6Clo/Ly6Chi比值下降(Figure 4A–4D、S8)。同时,血清IL-1β、IL-6和TNF-α水平升高,CD31+新生血管和I型胶原沉积均减少,表明炎症消退和早期组织修复均受损。

图4 CD40 缺陷会损害心梗后巨噬细胞向修复表型的转化
Result5 CD40-TRAF2/3/5 介导心梗后修复
CD40的两条下游通路中,TRAF2/3/5 介导心脏修复,TRAF6 主要介导促炎而非修复作用。
构建 CD40-TRAF2/3/5−/−与 CD40-TRAF6−/−基因敲入小鼠(Figure 5A),心梗 28 天后磁共振与超声显示,CD40-TRAF2/3/5−/−小鼠心室扩张更严重、射血分数与短轴缩短率显著降低,梗死与纤维化面积大于 CD40 KO 小鼠(Figure 5B–F);而 CD40-TRAF6−/−小鼠心功能、梗死及纤维化程度均接近野生型水平(Figure 5G–H)。

图5 心梗后 CD40-TRAF2/3/5 缺陷导致梗死面积扩大、心功能受损
Result6 CD40-TRAF2/3/5缺失损害巨噬细胞胞葬能力
CD40-TRAF2/3/5信号通路是调控巨噬细胞胞葬的关键下游通路,其缺失导致心肌修复障碍,CD40-TRAF2/3/5 通路是调控巨噬细胞胞葬的核心。
免疫荧光显示CD40 KO和CD40-TRAF2/3/5−/−小鼠心脏中TUNEL+凋亡心肌细胞显著增多,巨噬细胞吞噬凋亡心肌细胞的能力明显下降,而CD40-TRAF6−/−小鼠与WT小鼠相当(Figure 6A、6B)。。
腹腔注射FITC+酵母聚糖实验中,CD40-TRAF2/3/5−/−小鼠巨噬细胞吞噬能力最弱,而CD40-TRAF6−/−小鼠吞噬能力甚至优于WT(Figure 6E、6F)。体外BMDM实验进一步证实,CD40-TRAF2/3/5−/−巨噬细胞胞葬能力和CD206+修复表型极化均显著受损,MerTk和Arg1表达降低,而CD40-TRAF6−/−巨噬细胞与WT无显著差异(Figure 6G–6K)。
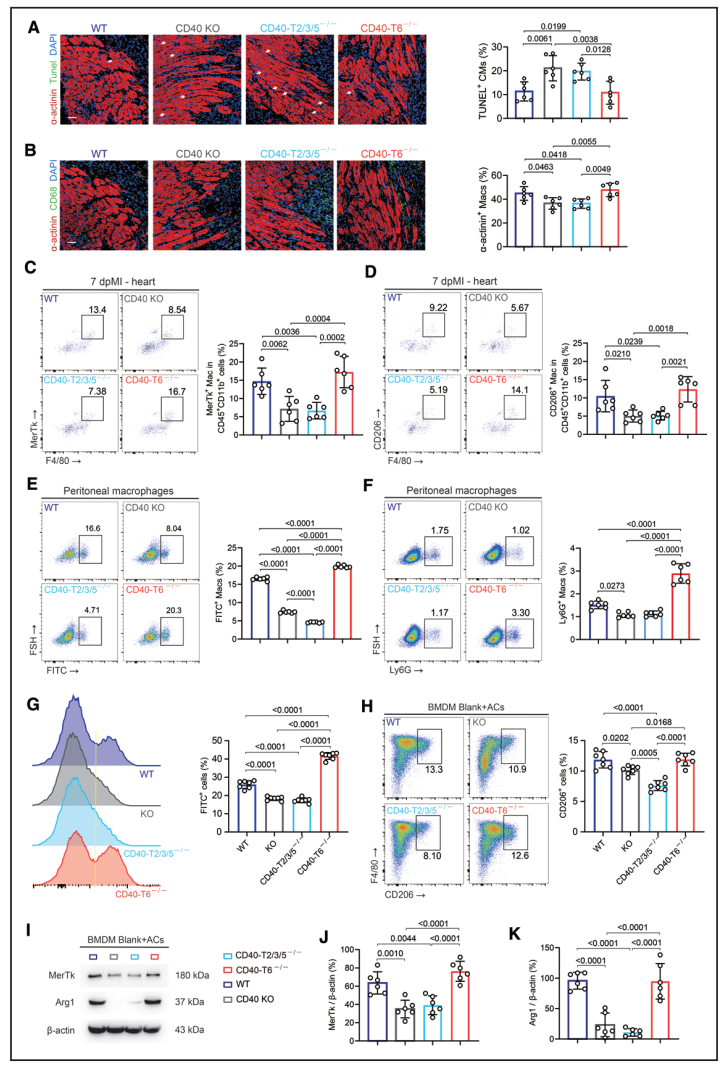
图6 CD40-TRAF2/3/5 缺陷(而非 CD40-TRAF6 缺陷)会损害巨噬细胞的胞葬作用
Result7 CD40-TRAF2/3/5 通过 STAT6 通路促进胞葬
STAT6 是 CD40-TRAF2/3/5 调控巨噬细胞胞葬的关键下游效应分子,CD40-TRAF2/3/5 通过激活STAT6通路上调MerTK与Arg1,增强胞葬。
蛋白质组与 Western blot 显示,CD40-TRAF2/3/5 缺失导致STAT6及其磷酸化水平显著降低(Fig. 7A–F)。
STAT6可直接结合MerTK、Arg1启动子并调控其表达(Fig. 7C–E)。
使用STAT6抑制剂后,TRAF2/3/5与TRAF6缺陷巨噬细胞的胞葬差异消失(Fig. 7K–L)。

图7 巨噬细胞 CD40-TRAF2/3/5 信号通过上调并激活 STAT6 促进胞葬作用
Result8 过表达ΔCD40可显著改善心梗后心脏修复
在巨噬细胞中特异性过表达保留TRAF2/3/5、缺失TRAF6的ΔCD40,可有效增强胞葬、缩小梗死、保护心功能。
腺病毒介导ΔCD40在巨噬细胞中特异性表达,转染效率与特异性得到验证(Fig. S19)。
ΔCD40过表达显著提高左室射血分数、减小心室容积、降低纤维化面积(Fig. 8B–F)。
ΔCD40提升 MerTK+巨噬细胞比例、增强凋亡心肌细胞清除、促进修复型巨噬细胞转化(Fig. 8G–J)。

图8 过表达 ΔCD40 可改善心梗后心功能与巨噬细胞胞葬作用
研究结论
本研究证实,巨噬细胞CD40通过TRAF2/3/5-STAT6通路促进胞葬作用,进而改善心肌梗死后心脏修复,而TRAF6通路主要介导促炎反应。研究颠覆了CD40仅为促炎分子的传统认知,明确了两条下游通路的功能分化。该成果揭示了心梗修复的全新免疫调控机制,提出选择性激活CD40-TRAF2/3/5是更安全有效的干预方向,为临床心肌缺血损伤治疗提供了创新靶点与理论基础,同时鉴定的修复型巨噬细胞前体也可为心梗预后评估提供新标志物。
参考文献
Linlin, Zhang,Yanshan, Chen,Pengpeng, Xu et al. CD40-TRAF2/3/5 Signaling Promotes Cardiac Repair by Mediating Macrophage Efferocytosis After Myocardial Infarction.[J] .Circulation, 2026, undefined: .