






前 言
单细胞RNA测序和ATAC测序是目前单细胞比较成熟的两种技术,可以分别研究细胞转录组状态和染色质可及性。2019年10月《Nature Biotechnology 》刊登的一篇在单细胞中同时进行RNA和ATAC测序的文章,将两种技术在同一细胞中进行了联合。具体的研究思路和结果是怎么样的呢?下面我们就来解读一下这篇高分文章。
基本信息
文章:High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell
期刊:Nature Biotechnology
影响因子:31.9
发表时间:2019.10.14
材料:小鼠的大脑皮层细胞
技术:SNARE-seq

背景介绍
单细胞RNA测序可以揭示细胞的转录状态,但对染色质这种上游调节领域研究较少。在同一个细胞内联合分析染色质和RNA 有助于理解转录调控及下游作用机制。作者推出一种基于液滴单细胞核染色质可及性和mRNA表达测序(SNARE-seq)的方法,可以将单个细胞转录组和它的可及染色质关联起来进行大规模测序。具体来说,使用Tn5转座酶在通透性细胞核中捕获可及性位点,在液滴中,使用DNA barcode标记来自同一细胞的mRNA。
为了证明SNARE-seq的结果有效性,作者分别从新生和成年小鼠的大脑皮层细胞收集了5081和10309个细胞。作者重建了主要和稀有细胞类型的转录组和表观遗传图谱,发现了谱系特异的可及性位点,特别是低丰度细胞,并将启动子可及性动态与神经发生过程中的转录水平联系起来。
补充知识
真核生物的核DNA不是裸露的,而是与组蛋白结合形成染色体的基本结构单位核小体,核小体再经逐步的压缩折叠,最终形成染色体高级结构(如人的DNA链完整展开约2m长,经过这样的折叠就变成了纳米级至微米级的染色质结构而可以储存在小小的细胞核)。而DNA的复制转录是需要将DNA的紧密结构打开,从而允许一些调控因子结合(转录因子或其他调控因子)。这部分打开的染色质,叫做开放染色质,打开的染色质允许其他调控因子结合的特性称为染色质的可及性(chromatin accessibility)。因此,认为染色质的可及性与转录调控密切相关。
研究方法
为了同时分析来自同一细胞核的染色质可及性和mRNA,作者推出了基于微液滴平台的SNARE-seq。在这个方法中,通透性细胞核中的可及性染色质在液滴形成前被Tn5转座酶捕获。作者之所以在液滴形成前捕获,是因为没有加热和洗涤处理,在转座后将转座酶结合到DNA的时候,可以保持原始DNA链的连续性,允许把同一细胞核的可及性基因组位点和mRNA共同包裹在同一个液滴中。作者设计了一个能够在5′与转座插入的接头序列互补,3′拥有被oligo(dT)-bearing barcoded 磁珠捕获的 poly(A) 碱基的splint oligonucleotide。在细胞核被液滴包裹之后,通过加热液滴,mRNA和碎片化的染色质被释放出来,以允许splint oligonucleotides和adaptor-coated beads与cellular barcode结合。然后对RNA和染色质可及性建库,用于测序(图1a )。结果数据可以通过cellular barcode进行关联,而不需要分别分析之后再进行单细胞clusters的映射关联。
研究结果
为了评估SNARE-seq 对可及性染色质的捕获效率,作者首先在具有广泛染色质特征的类淋巴母细胞系GM12878 进行了一个验证试验。结果表明SNARE-seq 可及性数据与ATAC-seq 和Omni-ATAC 有相似的信噪比(图1b)。整体的SNARE-seq数据也与之前文献报道的核小体模式一致,在典型启动子区有片段的强富集(图2ac),这是ATAC-seq数据的典型特征。作者对SNARE-seq peaks数据和已报道的ATAC-seq 和 Omni-ATAC peaks数据取交集(图2b),发现85.9% 的ATAC-seq peaks能够在另外两组中找到,87.6% 的Omni-ATAC peaks 可以在SNARE-seq 中找到。在过滤掉低质量数据后,作者获得的细胞可及性位点中位数为2,720,这与许多单细胞或者单细胞核ATAC-seq 文献报道的数量相当(图2d)。
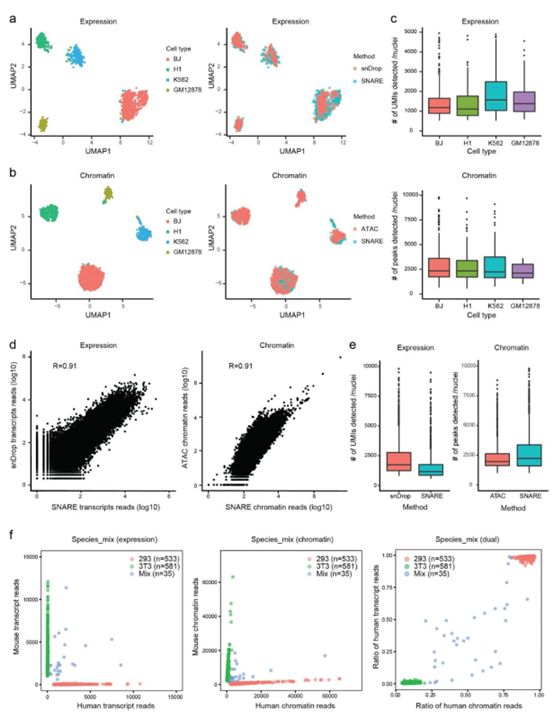
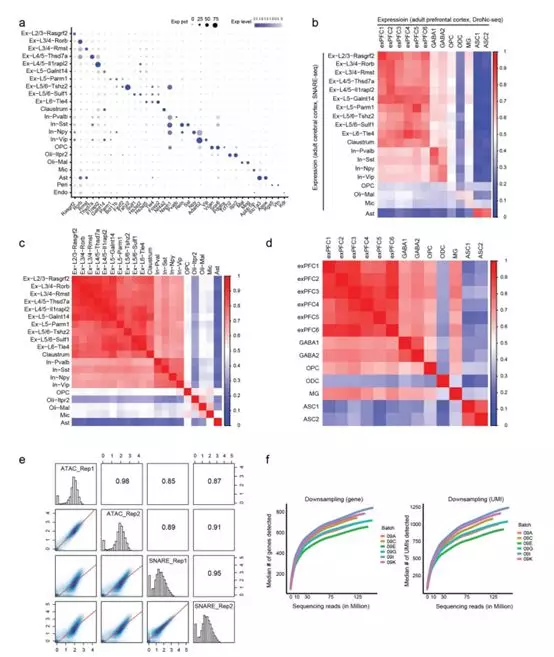
为了评估SNARE-seq 细胞类型鉴定的准确性,作者对人类BJ, H1, K562 和 GM12878 细胞的混合样本进行了SNARE-seq,收集得到1047对profiles (UMIs中位数为 500 ,可及性位点中位数为805)。分别进行的表达聚类和染色质可及性聚类鉴定得到四个明显区分的clusters(图1c)。差异表达的maker基因验证了以上聚类鉴定结果(图3a)。两对profiles 的类型鉴定结果有很好的一致性(kappa coefficient 0.92, 图1d)。需要特别指出的是,作者发现转录因子JUN,IRF8,POU5F1 和 GATA1分别在BJ, GM12878,H1 和 K562 细胞中富集表达(图3c),SNARE-seq可行性分析表明这些转录因子优先结合到染色质的模式比较相似,这与之前的观察一致。作者为了提高检测的灵敏度,使用NP-40-based Nuclei EZ buffer提高标记效率,添加一种RNA酶抑制剂以防止RNA的降解。作者在细胞混合样本中获得1,043对profiles (UMIs中位数为 1159 ),中位数为2254的可及性位点(图4c)。作者比较了SNARE-seq 两组学数据和snDrop-seq、SNARE-seq chromatin-only数据,发现三者聚类结果具有一致性(图4ab),raw reads的相关性水平较高(图 4d),回收效率也比较高(图 4e, 66% 的RNA回收率, 100% 的染色质回收率)。此外,物种混合实验表明SNARE-seq数据的高纯度和低倍增率(6% ,图 4f)。因此,在一个简单的混合细胞中,SNARE-seq能在染色质和转录组基础上有效分离细胞类型,且具有高度的一致性。
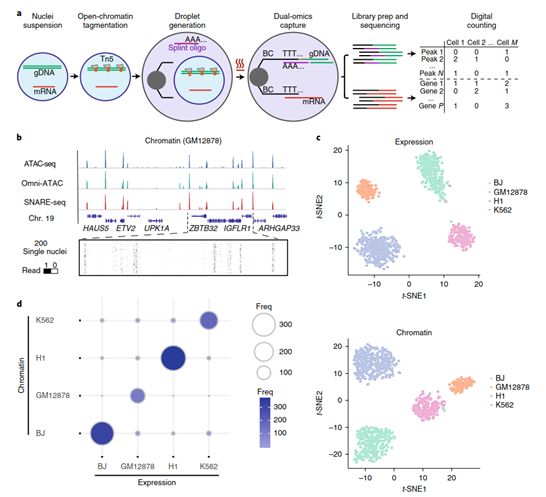
图1 | 人单细胞核转录组和染色质可及性联合测序
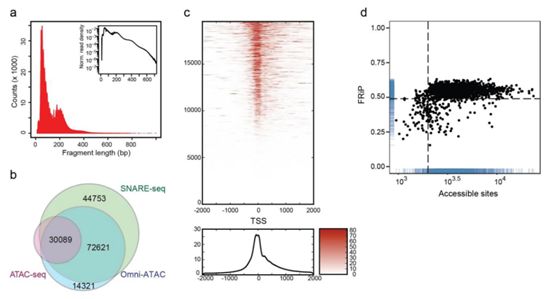
图2 | GM12878细胞SNARE-seq染色质谱的质量测定
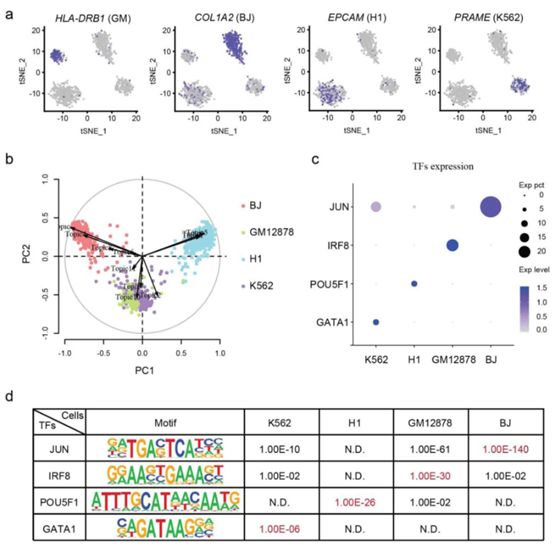
图3 | 人类细胞系混合样本SNARE-seq 细胞类型鉴定
作者接下来使用SNARE-seq对小鼠新生大脑皮层进行分析,质量过滤之后获得5,081细胞核,其中转录组UMIs中位数为357,染色质可及性位点中位数2,583。转录组表达谱和蛋白质谱相关性表明双组学对单一组学的SNARE-seq重现度较高(图5ab)。在所有的RNA reads中,94% 比对到基因组, 37% 比对到外显子 ,42% 比对到内含子(图5c),反映了新生期转录本在细胞核中的富集情况。相比之下,尽管比对率差不多(>91%),染色质可及性数据中比对到基因间区的reads比例更高。可及性reads在接近转录起始位点的区域富集,在外显子区覆盖度较低,表明在这部分非编码区中存在着增强子和启动子序列。因此,相较于snDrop-seq 和 snATAC 数据,RNA和染色质reads在基因组上的分布与预期一致。
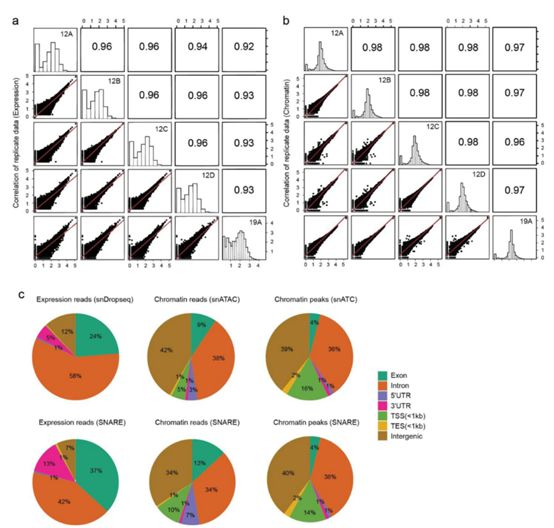
图5 | SNARE-seq 的重现性(n=5 replicates)
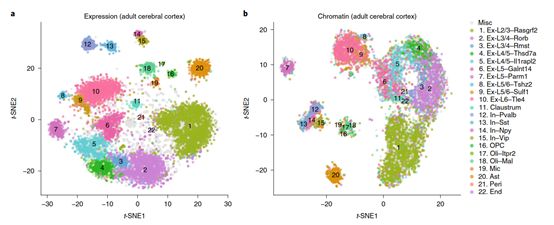
大脑皮层转录组的非监督聚类鉴定得到19个细胞clusters,包括星形胶质细胞/放射状胶质细胞(Ast/RG)、中间祖细胞(IP)、兴奋性神经元 (Ex)、迁移抑制性神经元(In)和Cajal–Retzius 细胞。作者进一步检测到几种非神经元细胞类型,包括少突胶质细胞祖细胞(OPCs)、内皮细胞(End)、周细胞(Peri)和小胶质细胞(Mic)。这些细胞clusters的细胞数目从37 (0.7%) 到542 (10.7%) 不等(图6a),且与批次和测序深度无关(图6b-e)。UMAP结果展示了一个从祖细胞开始延伸的轨迹,反映了皮质细胞发育的顺序。新生的浅层神经元和神经胶质细胞类型与在这个时间点的胶质生成的起始有关(图7a)。作者对SNARE-seq转录组数据和最近发表的基于SPLiT-seq的小鼠相似发育时期的皮层单细胞RNA-seq数据集进行了比较,尽管SNARE-seq UMIs数量偏低,但在细胞类型和显著特征上表现出了相当好的相关性(图8a–c)。值得注意的是,作者发现了中间祖细胞紧密相关的细胞状态之间的细微差别,并鉴定得到三种细胞亚型:代表循环祖细胞的cluster IP–Hmgn2,表达Mki67,Top2a和Kif23(图7b8b),代表退出细胞循环的顶端祖细胞的cluster IP–Gadd45g中Gadd45g高表达, 代表基底祖细胞的cluster IP–Eomes,这些clusters的细胞类型和细胞层鉴定在接下来的makers 基因表达和markers原位杂交染色中得到验证(图7b,图9)。
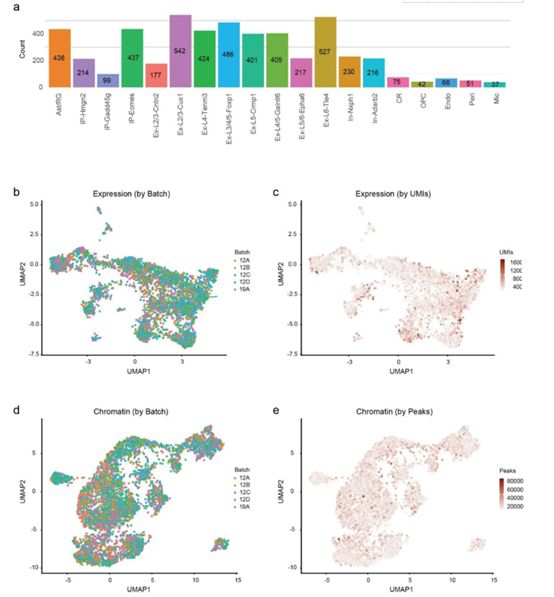
图 6 | SNARE-seq的稳定性
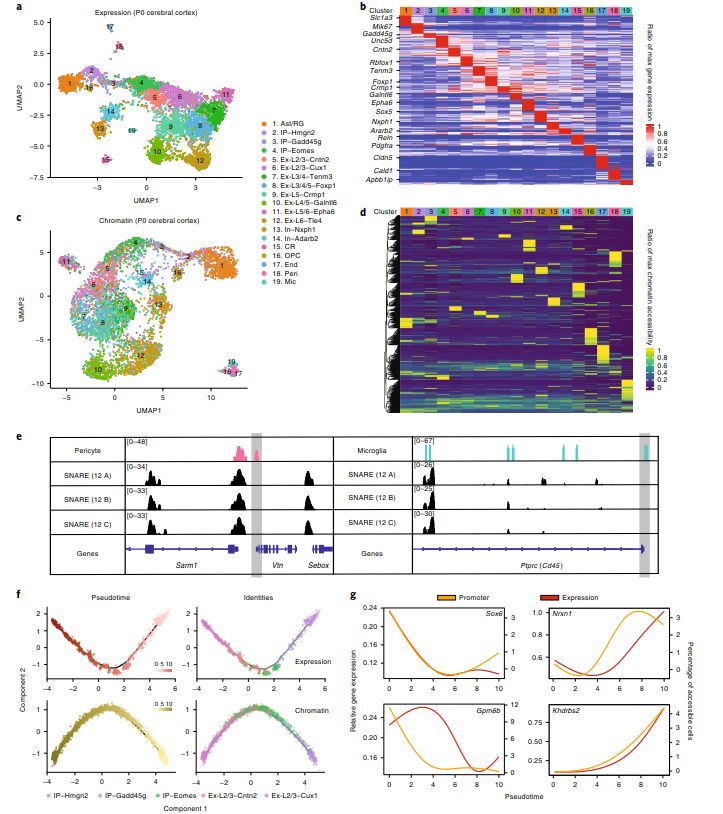
图7 | 新生小鼠大脑皮层SNARE-seq双组学分析 (n = 5 replicates)
作者对新生小鼠大脑皮层SNARE-seq 染色质可及性profiles和已发表的ATAC-seq ENCODE数据进行了比较,发现两者之间存在高度的一致性(图8ef)。为了基于染色质可及性数据对共有细胞进行聚类,作者使用相应的转录组表达谱分别合计每个cluster的染色质可及性信号,然后进行peak calling以及cisTopic中的概率主题建模方法(probabilistic topic modeling method)分析。使用UMAP降维之后,大多数单核染色质可及性clusters (图 8c)与相应的转录组表达获得的细胞类型一致(图8a)。值得注意的是,染色质可及性结果中,处于大脑皮层和神经节分化早期的深层兴奋神经元和迁移抑制性神经元能够被很好地聚类区分,而晚期生成的浅层兴奋性神经元则区分地不太明显。基于转录组表达不能清晰区分的边界部分,在染色质信息的基础上可以被进一步聚类为某些细胞亚型(图10ab)。通过loci Hes5 (Ast/RG),Gadd45g (IP),Meg3 (neuron),Pdgfra (OPC),Vtn (Peri) 和 Apbb1ip (Mic)这些启动子区可及性特异的marker 基因对主要clusters进一步进行细胞类型鉴定(图10d),作者发现 Vtn 和Cd45的启动子可及性仅存在于使用转录组数据de novo鉴定得到的cell-type-aggregated染色质谱中。相反,基于batch-aggregated profiles获得的可及性peaks,使用现有默认策略进行sc-ATAC-seq 数据分析,不能在其他高丰度细胞产生的背景噪音存在的情况下重现稀有细胞特有的可及性peaks(图7e)。与此结论一致,没有任何表达数据的蛋白质谱聚类只能获得不太清楚的细胞类型边界,许多低丰度的细胞类型无法被检测到(图10 c)。所以,关联了基因表达谱的染色质可及性数据的细胞类型鉴定结果可以更灵敏地检测可及性染色质区域。
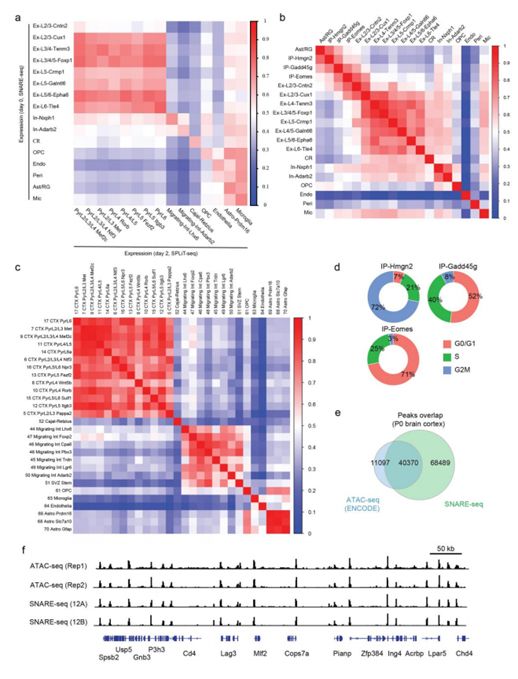
图8 | 新生小鼠大脑皮层SNARE-seq结果与已发表的表达和染色质数据具有相关性
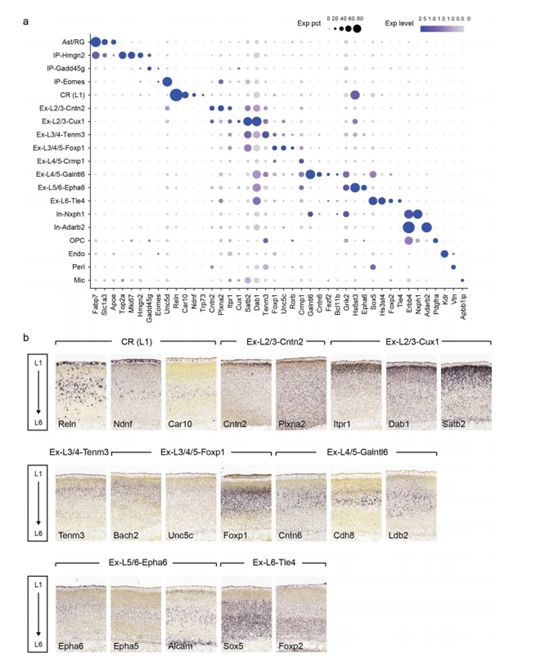
图 9 | 小鼠新生大脑不同细胞类型marker基因表达数据
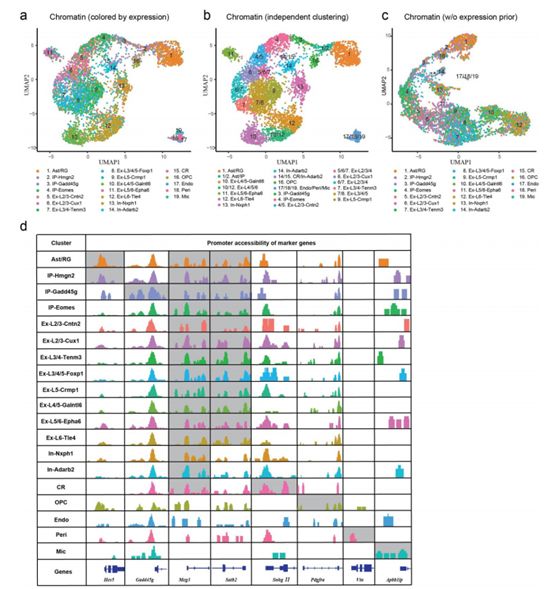
图 10 | 新生小鼠大脑皮层SNARE-seq 单细胞染色质可及性聚类 (n=5,081)
接下来,作者重点研究了从中间祖细胞到上层兴奋神经元的发展情况。作者使用Monocle,基于top差异基因(qval < 0.05,图7 f),对1,469个核基因表达谱进行了拟时序分析。从转录动态中,作者发现一个起源于cell-cycle-exited期的从成神经细胞期(Eomes and Unc5d)到Foxp1-expressin 和Cux1-expressing的上层神经元的清晰模式(Mki67 and Gadd45g)(图11a),基于相同细胞核的1,332可及性位点数据集,作者进一步确定了差异可及性的另一个轨迹(图7f下面部分)。这些不同的发育轨迹说明了拟时序之间的高相关性。在这些差异可及性位点中,启动子区有103个,21个相关基因在拟时序中差异表达。其中大部分基因启动子可及性和表达水平上表现出相似的趋势变化(图11bc)。例如,Sox6 和Mlc1在神经分化期表现出下降趋势。而Khdrbs2 (SLM1)以及他调控的靶基因Nrxn1,在神经发生中呈现出相似的上调趋势(7g 和11c,d)。SNARE-seq连锁表达和蛋白质可及性profiles可以构建发育过程中的调节动态,并能详细描述细胞类群的表观遗传状态(图12)。
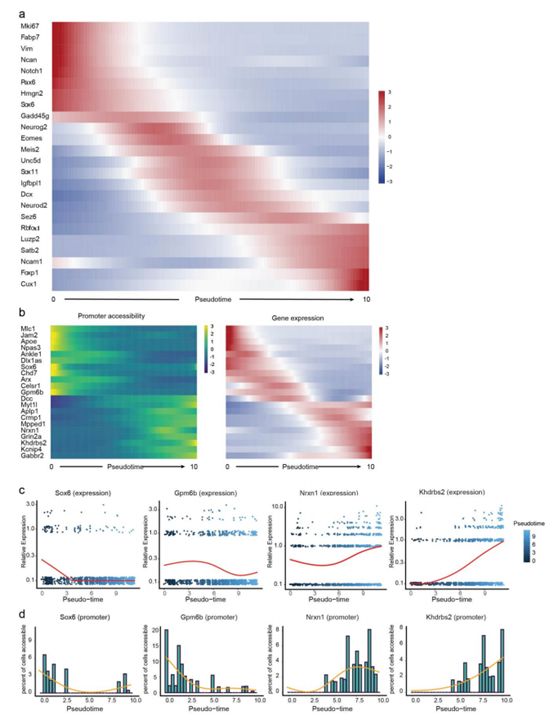
图 11 | 拟时序分析早期神经发生阶段的基因表达动态过程
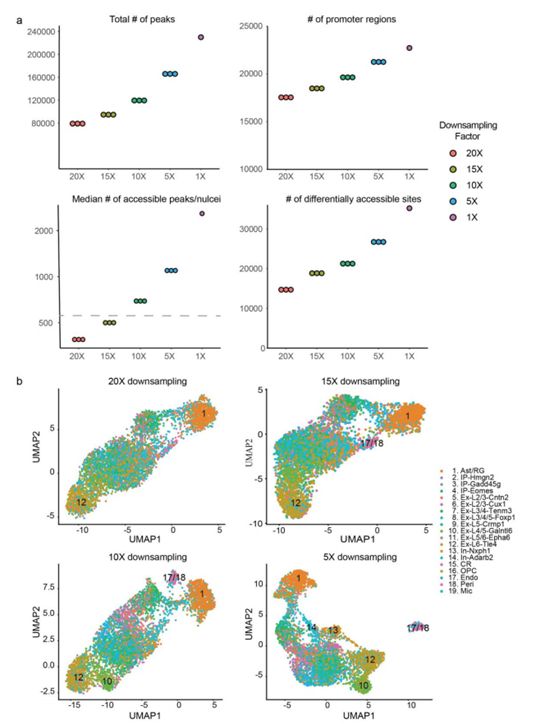
图12 | SNARE-seq 染色质数据的敏感度
最后,作者将SNARE-seq应用于成年小鼠大脑皮层组织,质控过滤后获得10,309对profiles(RNA UMIs 中位数1,332,染色质可及性位点中位数2,000)。对这10309 转录组数据进行非监督聚类获得22个细胞clusters,包括10种兴奋性神经元,4种抑制性神经元(表达Pvalb,Sst,Npy 和 Vip),OPCs,新形成的表达Itpr2的少突胶质细胞(Oli–Itpr2),成熟少突胶质细胞(Oli–Mal),以及其他的非神经细胞(图13a)。clusters中大多数可以通过现有谱系或者皮层markers进行鉴定。这些markers基因表达相似但有着更多的特异性(图9 和图14a–d)。为了研究每个细胞cluster的表观遗传模式,作者分析了成年小鼠大脑皮层SNARE-seq的染色质数据,发现在相关转录组数据de novo聚类鉴定得到的细胞类型方面,和ATAC-seq数据结果十分相似(图14e)。相比于新生大脑皮层染色质profile鉴定得到的细胞clusters,作者使用the topic modeling method进行peak calling 和聚类得到的clusters更加干净并且显著区分,可能是因为成年大脑细胞状态本身比较离散。作者又对所有细胞clusters的差异可及性peaks 进行基因功能分类和motif富集分析,相比较于发育期小鼠的大脑皮层,虽然有些clusters,比如星形胶质细胞(Ast)和小胶质细胞(Mic)表现出相似的生物过程和转录因子结合富集,但大多数clusters呈现出显著的特征差别,这可能反映了出生后大脑的成熟过程。
总结
SNARE-seq是一种可以同时分析单细胞或者单细胞核转录组和染色质可及性的稳定方法,因为其中试剂的简单设计,SNARE-seq可以得到更广泛和有效的应用。与其他单细胞核RNA-seq方法相比,SNARE-seq在检测RNA 方面更加灵敏(图7de和图14f),并能捕获多达4-5倍的可及性位点(图7ab),提高了大约两倍的差异可及性位点鉴定率,并且使细胞clusters更好地分离(图13)。最后,SNARE-seq的通量可以通过细胞组合索引(cellular combinatorial indexing)方法进一步提高。SNARE-seq是一个包括转录调节单元上下游在内的组织复杂性特征描述的有效工具,尤其可以用于构建人类组织和临床样本的细胞图谱。
参考文献
Chen S., Lake B., Zhang K., High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nature biotechnology (2019): 1-6.