






合理的参考基因组版本对转录组项目分析尤为重要。随着高通量测序技术的发展,物种的参考基因组版本越来越多,那么,同一物种不同版本参考基因组该如何选择呢?今天小编来详细介绍一下在做特定物种的转录组测序前,如何进行参考基因组的查找和评估。
如何查找?
常用的数据库主要有NCBI、Ensembl和JGI三种数据库。下面我们以模式物种小鼠(Mus musculus)为例说明。
一、NCBI数据库
NCBI(National Center for Biotechnology Information)是美国国立卫生研究院(NIH)的国立医学图书馆(NLM)的一个分支。NCBI检索系统收录了许多基因序列和蛋白序列数据库。具体查找操作步骤如下:
1、打开NCBI(https://www.ncbi.nlm.nih.gov),选择下拉条目“Genome”并在搜索框中填写所需要查找基因组的物种(Mus musculus[orgn]),点击搜索。
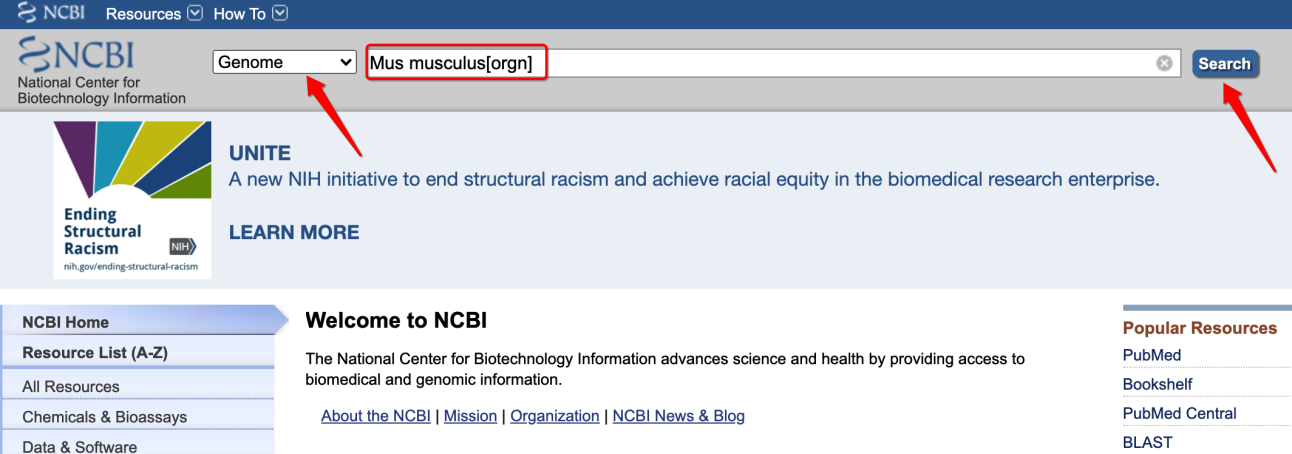
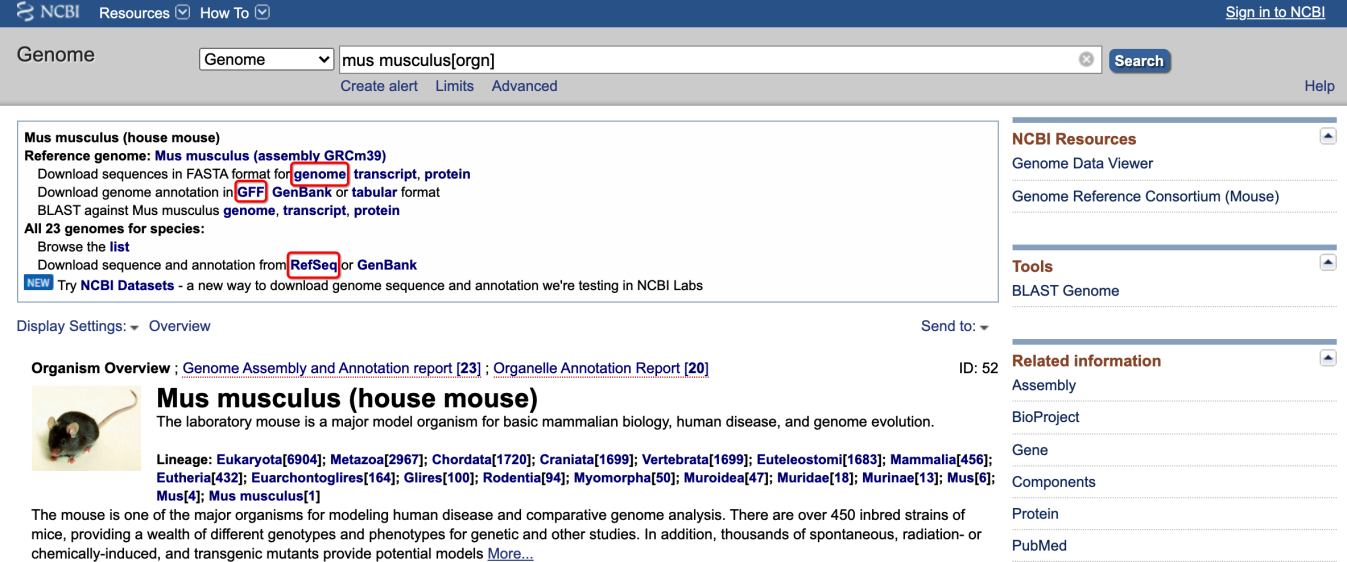
2、下图框定的内容为评估物种参考基因组需要的信息文件,点击即可下载。
二、Ensembl数据库
Ensembl是由 European Bioinformatics Institute(EBI)与Wellcome Trust Sanger Institute(WTSI)共同合作开发的数据库项目。涵盖大量物种的参考基因组信息,并且数据更新及时。具体查找操作步骤如下:
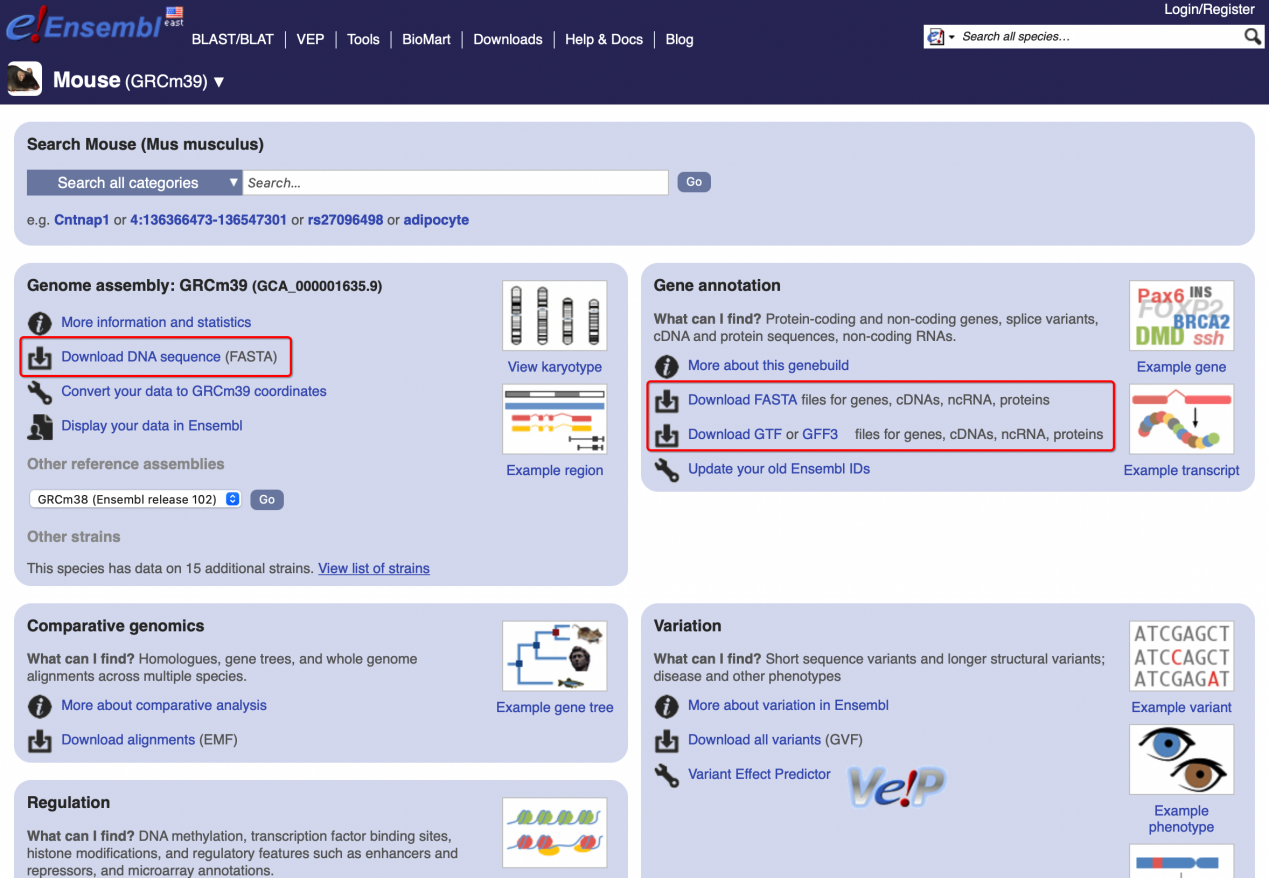
1、打开Ensembl(http://www.ensembl.org/index.html )网站主页, 找到标注为“All genome”的物种选项。若物种为植物,则对应网站为Ensembl Plant(http://plants.ensembl.org/index.html)。
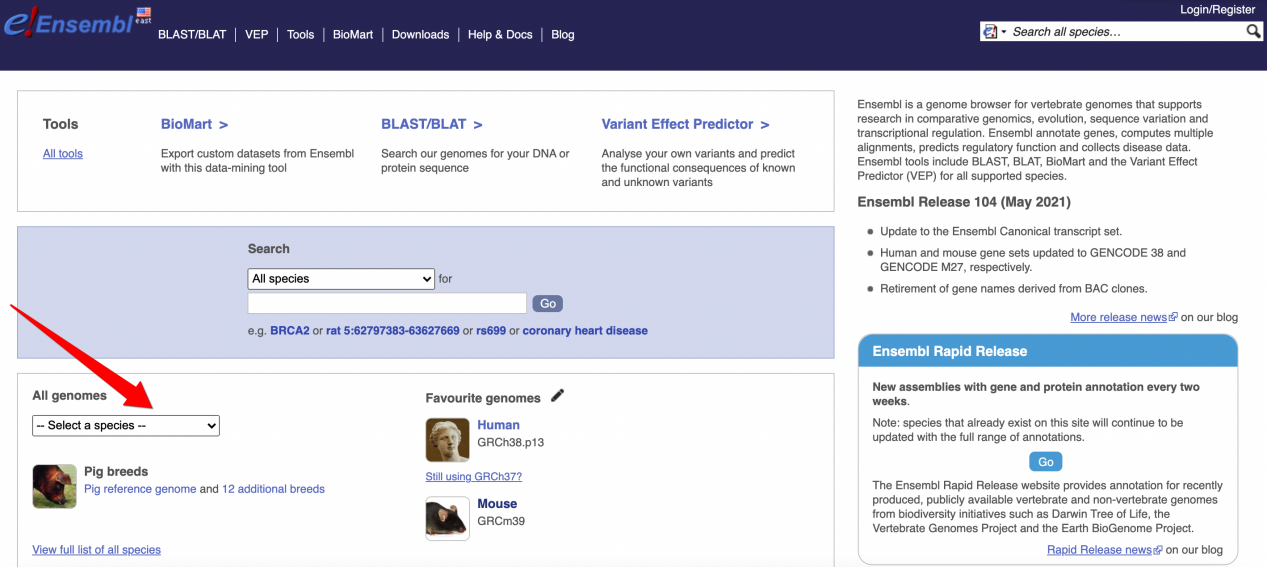
2、如图所示位置选择需要下载的物种。
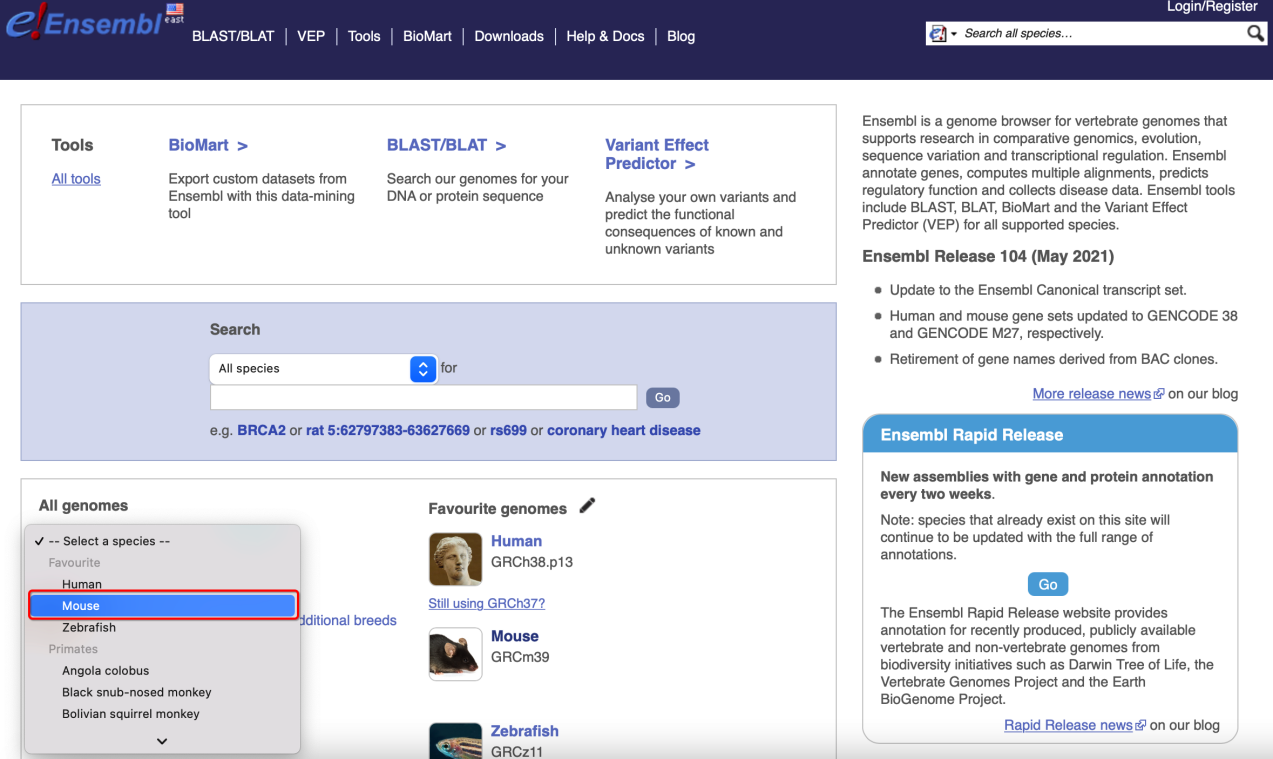
3、下图框定的内容为评估物种参考基因组需要的信息文件,点击下载即可。
三、JGI数据库
部分植物和真菌基因组可在JGI数据库(https://genome.jgi.doe.gov/portal/)中查找。
四、其他数据库
1、GigaDB数据库(http://gigadb.org/#myCarousel)
2、国家基因组生命大数据库(https://db.cngb.org/)
3、plaBi数据库(https://www.plabipd.de/index.ep)
此数据库里面记录了已测序了的植物以及发表的文章。根据植物分类来查找具体发表的文献。
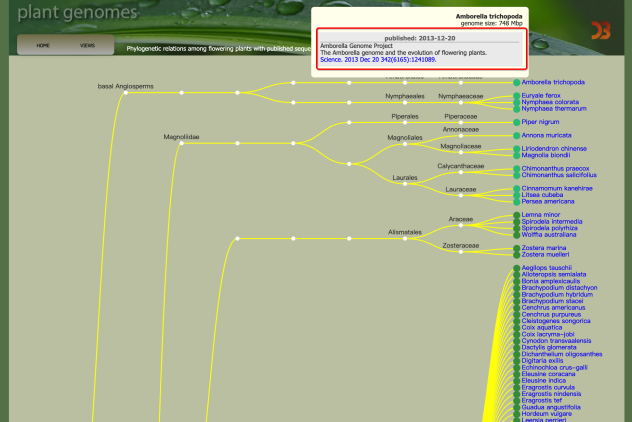
如何评估?
在掌握了参考基因组查找下载方法之后,接下来我们详细介绍下载后的参考基因组信息的评估方法。
一、组装指标
1、组装水平
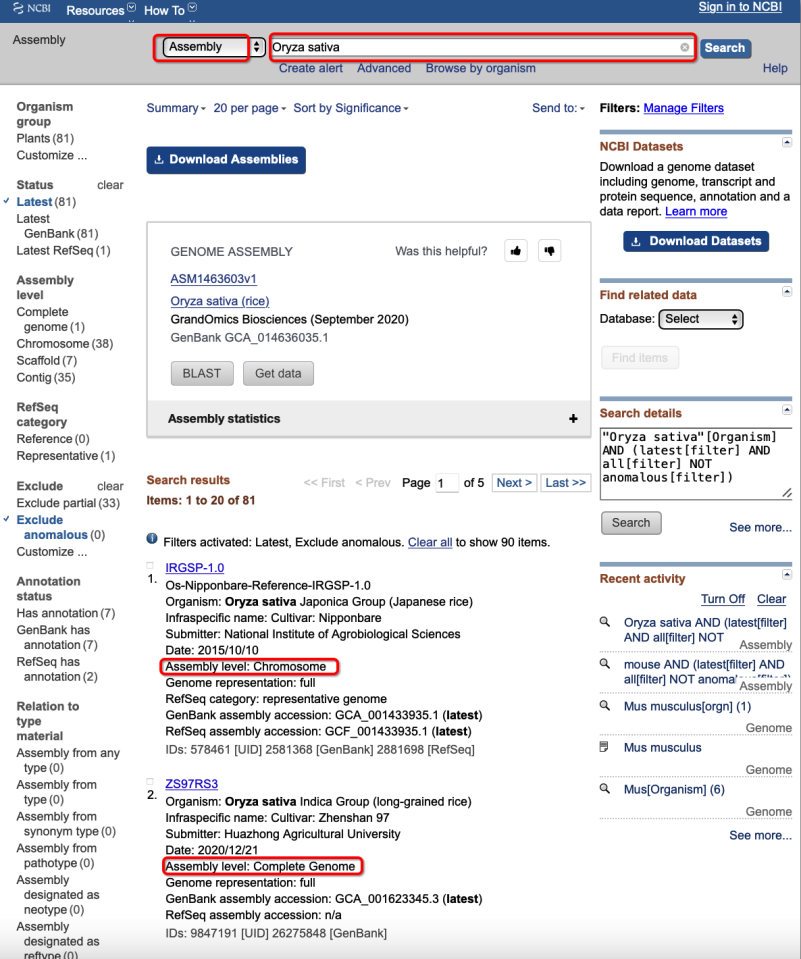
以水稻(Oryza sativa)基因组为例,在首页下拉条目选择“Assembly”并在搜索框中填写物种名称进行搜索。跳转页面会出现不同版本基因组的组装水平,一般来说组装水平越接近染色体组装效果越好(Complete Genome > Chromosome > Scaffold > Contig)。
2、组装方法
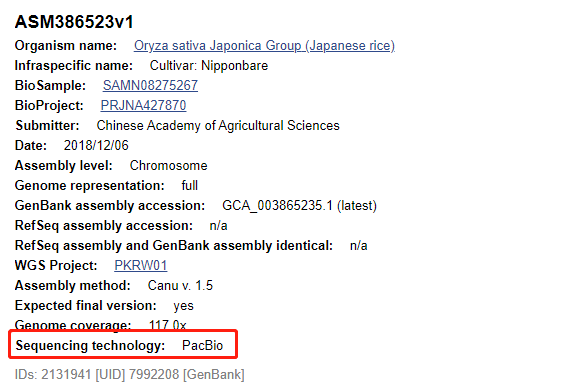
对一条染色体进行测序,将测序得到的reads进行拼接,能够完全拼接起来,中间没有gap的序列称为contig。将测序得到的所有contig从大到小进行排列,当其长度达到染色体长度的一半时,这一条contig的长度就叫做contig N50。我们可以用这个数值评估序列组装质量,值越大,组装效果越好。2018年以来,随着Pacbio 和 Oxford Nanopore Technologies(ONT)新技术的兴起,大大提高了基因组组装质量。一般来说,运用以上两种技术组装出的基因组较好(contig N50 > 1Mb)。
二、GFF/GTF文件评估
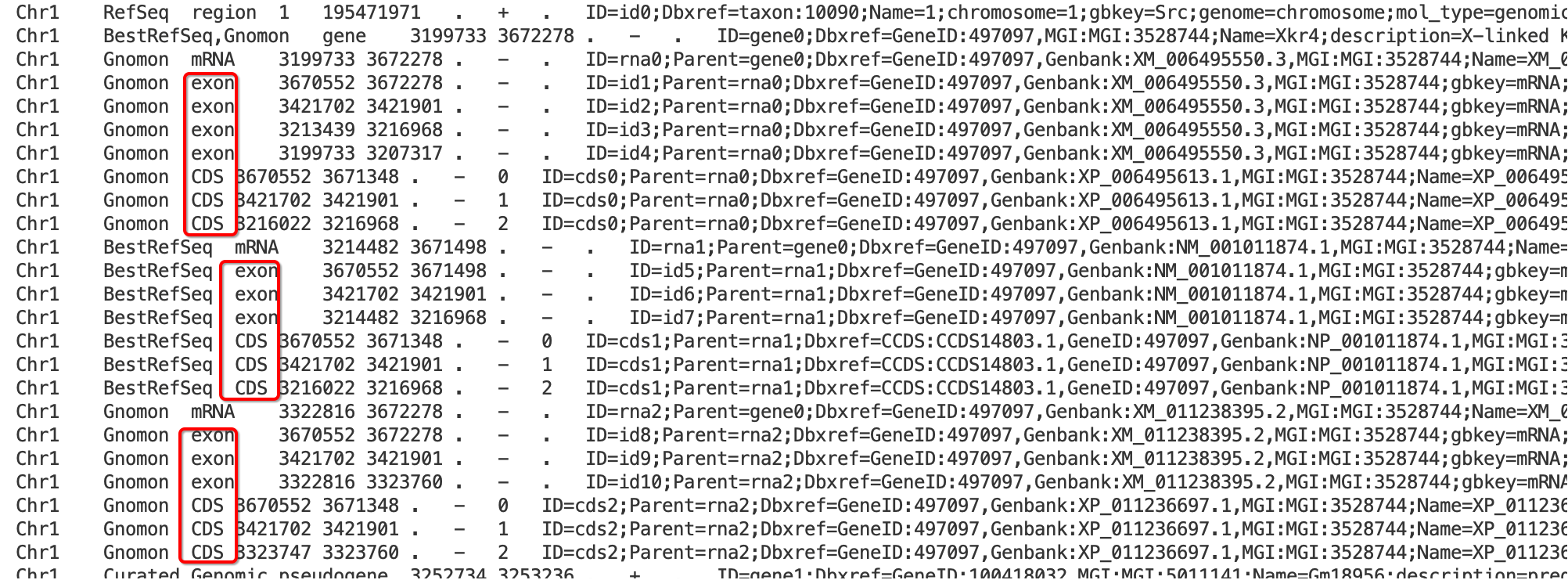
GFF(general feature format)主要是用来注释基因组。而GTF(gene transfer format)主要是用来对基因进行注释。下载GFF/GTF文件,用Notepad软件(https://notepad-plus-plus.org/downloads/)打开。仔细检查GFF文件,若显示有exon和CDS信息,则基因组注释相对完全,可用于分析;若exon和CDS缺失,说明该基因组注释不完整,需等完整注释信息进一步完善后才可分析。
三、序列一致性评估
通过reads比到基因组上,验证reads对基因组的覆盖情况,用于评估组装的完整性以及测序的均匀性。较高的mapping rate(90%以上)认为组装结果和reads有比较好的一致性。如果物种仅有genome,没有transcript和GFF文件,表明只有基因组序列,无法做完整常规有参, 一般是先做基因组比对,比对率达到70%以上,基于比对到基因组上的序列进行转录本组装,对组装的转录本进行注释,定量,差异和富集分析。
以上为查找和评估的参考方法建议,项目执行以生信分析工程师综合评估版本为准。