






前言
许多生物系统(例如胚胎,肝小叶,肠绒毛和肿瘤)的功能取决于其细胞的空间位置结果。在过去的十年中,科学家们已经开发出多种高通量实验技术来量化空间位置中的基因表达,同时也开发了各种利用空间基因表达数据来识别具有空间表达模式的基因并描绘组织内邻域结构的计算方法。为了评估空间基因表达技术驱动生物学发现的能力和潜力,该文提供了一个历史性的空间转录组学文献数据库,该数据库最早可以追溯到 1987 年,对领域的趋势进行了详尽的分析,例如使用的实验技术、研究的物种和组织以及使用的方法等。该分析文档将当前流行的技术方法置于历史背景下,从而获得了可以指导当前研究策略的领域见解。
1. 发展历程
1.1 前传时代
一些实现空间转录组学的重要技术,最早可以追溯到二十世纪七十年代,长期以来,各种形式的原位杂交技术(ISH)已用于可视化空间中的基因表达。放射性 ISH 于 1969 年首次引入,实现了爪蟾卵母细胞的 rRNA 和 DNA 可视化,并在 1973 年首次用于可视化特定基因(珠蛋白)的转录本。非放射性荧光或显色原位杂交(FISH)于二十世纪七十年代至八十年代初开发出来,其提高了空间分辨率,支持3D染色,并缩短了所需的曝光时间(图1A)。早期原位杂交在组织切片中进行,难以应用于囊胚和重建 3D 组织结构。整体原位杂交(WMISH)于 1989 年在果蝇中首次引入,并在二十世纪九十年代初应用于其他物种,例如小鼠。
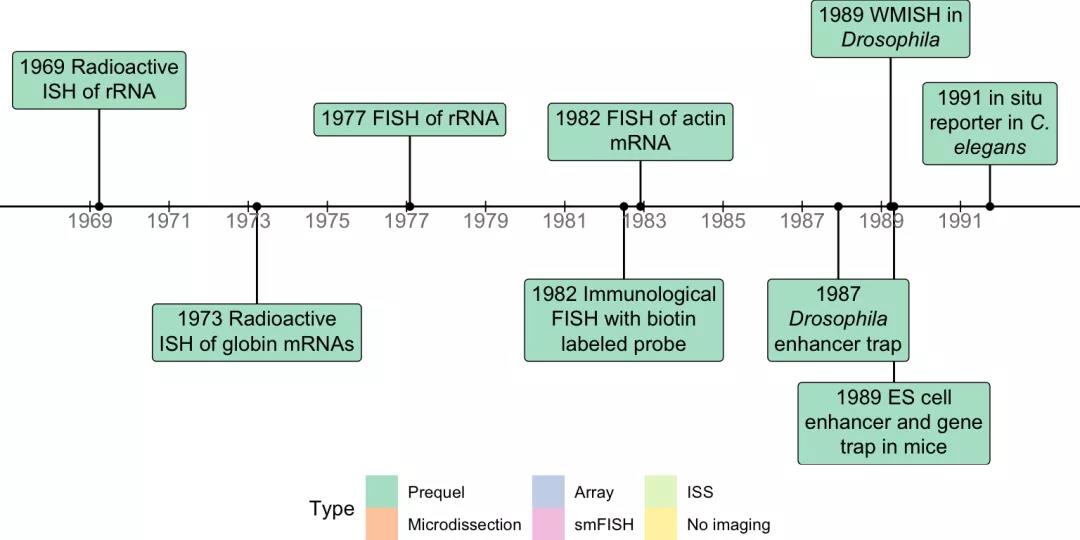
图1 前传时代空间转录组技术发展路线一
早期空间转录组学的另一条发展路线是基因捕获和增强子捕获筛选,开发于二十世纪八十年代,当时 DNA 测序通量正在增加,动物基因组是新开放的前沿领域。果蝇和小鼠的首次筛选是在八十年代后期进行的,目的是观察未靶向且通常未知的基因表达情况,可以通过对 cDNA 的 5' 末端快速扩增,对 PCR 产物进行 Sanger 测序来鉴定。
另外一个相关的技术--基因融合,通过报告基因与目标基因的预定调控序列融合,报告基因的表达模式能够概括目标基因的表达模式 。该技术于 1991 年首次在秀丽隐杆线虫的基因表达筛选使用。随着通量的增加,增强子和基因捕获技术在九十年代成为空间转录组学的技术,直到 WMISH 在九十年代后期兴起。WMISH 技术的自动化程度较高,避免了对转基因品系的依赖,同时由于在二十一世纪初获得物种的参考基因组信息变得越来越便利,探针设计因此也更为方便。

图2 前传时代空间转录组技术发展路线二
1.2 当前时代
许多当前时代空间转录组技术的基础都是在自七十年代以来的几十年间建立的(图3)。例如,1976 年首先使用紫外线激光来切割组织。流行的红外(IR)和紫外线(UV) 激光显微切割(laser capture microdissection,LCM)系统最早在 1996 年被报道,并很快商品化。微阵列技术于 1995 年首次报道,最初用于定量与固定在载玻片上的 cDNA 杂交的转录本,但很快在 1999 年被应用于定量 LCM 样品的转录组。当前流行的技术,例如空间转录组学(ST)和 10X Visium 依靠这种微阵列技术,区别在于是从放置在微阵列载玻片上的组织上而不是从溶液中来捕获转录本信息。一些通过单分子荧光原位杂交(smFISH)的高度多重化成像技术(例如 seqFISH)依赖于组合标签, 即利用多条短的寡核苷酸探针(大约20 bp)来靶向同一 mRNA 转录本的不同区域,每条寡核苷酸仅与一个荧光团偶联,因此本身的信号微弱。不过,多条寡核苷酸与同一转录本结合,就产生了可见的荧光点,并且提高了特异性,因此可以同时量化更多的基因或者转录本。组合标签于 1989 年在免疫 DNA FISH 中首次报道,并于 2002 年首次用于转录本;1998 年首次报道了 smFISH 的明确技术方法,显示每个 mRNA 分子都为斑点;没有这些技术的发展,就不可能实现高度复用的 smFISH。
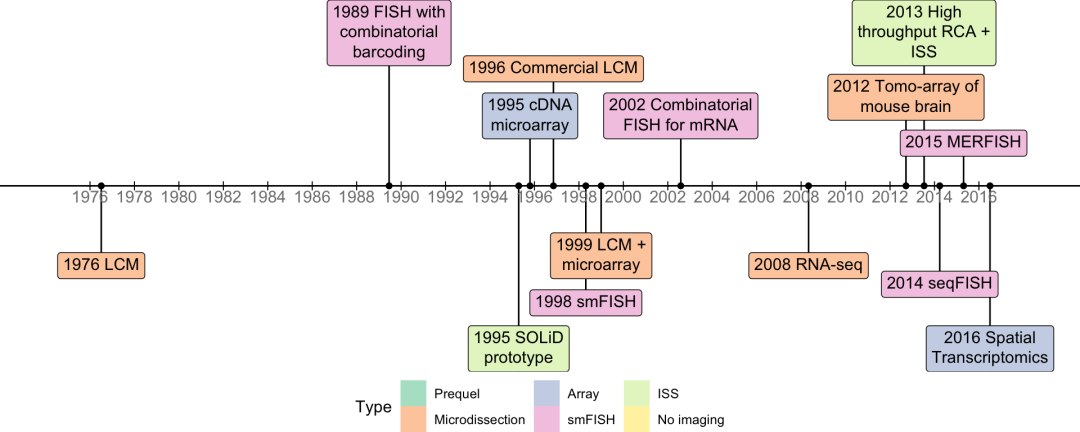
图3 当前时代空间转录组技术发展路线
2. 当前技术介绍
当前时代的空间转录组技术大致分为五类方向:激光显微切割(laser capture microdissection,LCM),单分子荧光原位杂交(single molecular fluorescent in situ hybridization, smFISH),靶向原位测序(In situ sequencing,ISS),原位阵列捕获(In situ array capture, Array) 和 其他非成像技术(No imaging)。
2.1 技术简介
激光显微切割
自1999年以来,使用最广泛的显微解剖技术是 LCM,它已应用于各种生物学领域,例如肿瘤学、神经科学、免疫学、发育生物学和植物学。在 UV LCM 中,UV 激光消融选择区域周围的一小段组织,然后通过重力或激光压力弹射器将其收集到试管中(图4 B,C)。在 IR LCM 中,组织切片安装在管帽上的塑料膜上,红外激光短暂加热所需区域使膜融化,使该区域的组织融合到帽上并被捕获(图4 A)。
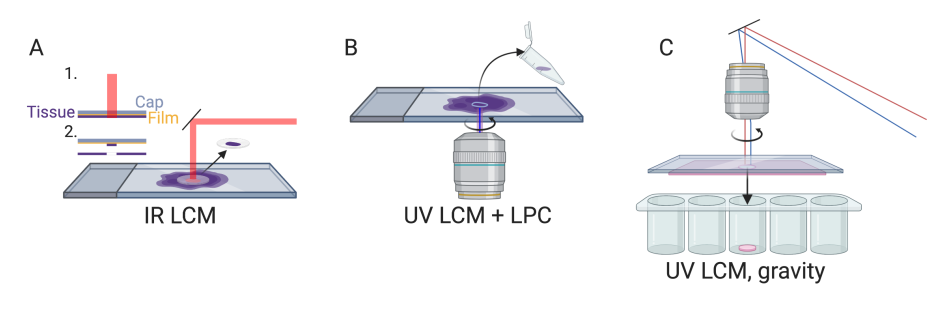

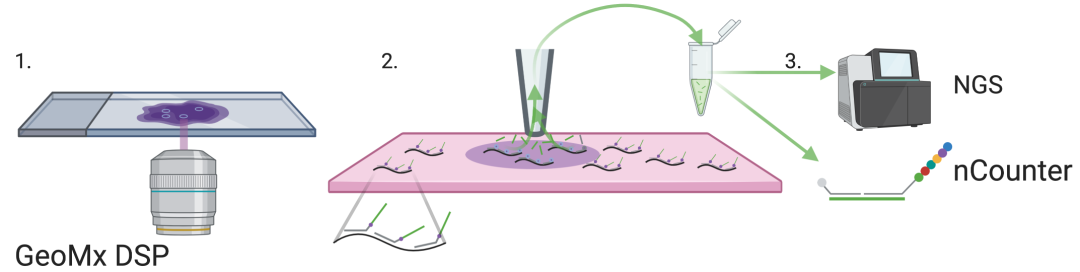
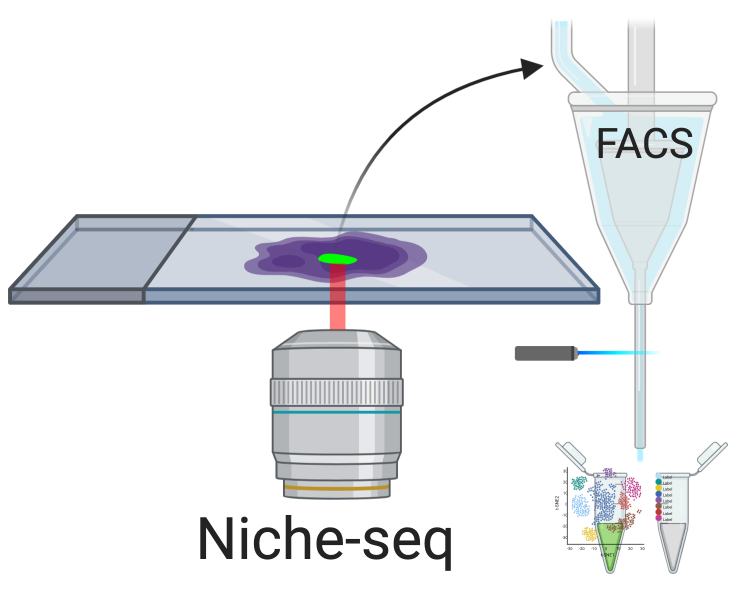
图4 显微切割原理示意图
空间转录组相关的显微解剖技术通常分为两类:机械和光学。前者包括 Tomo-seq 等,通过沿感兴趣轴,利用冷冻切片机进行组织切片,然后在每个切片上均进行 RNA 测序。光学显微解剖包括:GeoMX DSP,其将紫外线照射到感兴趣区域(ROI)上,释放可光裂解的基因标签以进行测序定量;Niche-seq,其使用荧光激活细胞分选(FACS)在转基因小鼠中分离具有光活化的 GFP 细胞,并进行 scRNA-seq 测序。
LCM 的优点包括能进行转录组整体水平分析,组织学切割精确,兼容福尔马林固定石蜡包埋(FFPE)组织等。LCM 也可以通过连续切割获得不重叠的区域,应用于 3D 组织重建,如 GEO-seq。LCM 的缺点则为操作复杂导致难以同时处理大规模样本,潜在的 RNA 降解风险等。
单分子荧光原位杂交
按时间顺序,当前时代开发的下一项技术是 smFISH,通过将单个转录本显示为带有 FISH 的不同点,对它们进行计数,从而对转录本丰度进行估计。代表技术之一是 seqFISH。在 2014 年发表的 seqFISH 中,为每个基因设计了二十四个单标记探针,并用 12 种基因编码了 4 种颜色和 2 轮杂交(图5)。在对第一轮杂交和 DNA 的 DAPI 染色进行成像后,用 DNase I 除去探针,然后进行第二轮的探针杂交。在实践中,会执行一轮错误校正杂交,因此,如果缺少一轮杂交的信号,仍然可以区分基因 。最近在 seqFISH+ 中,“颜色”本身是由杂交会产生热成像的条形码组成,该技术是将调色板扩展到 20 种“颜色”,能够分析 10,000 个基因。
另一种 smFISH 技术是 MERFISH,这是一个高度多重化的 smFISH 成像方法,可以在单个细胞中鉴定数千种 RNA 的拷贝数和空间定位, 该技术使用组合标签、连续成像等技术来提高检测通量,还通过 error-robust 编码方案来抵消单分子标记和检测错误。其他大多数基于 smFISH 的技术,例如 HybISS 和 split-FISH,则使用类似 seqFISH 或类似 MERFISH 的组合标签。
基于 smFISH 技术的优点包括基因的检测效率高、达到单细胞分辨率以及能够亚细胞定位的转录本。单轮 smFISH 具有近 100% 的检测效率,多轮杂交往往会降低效率,部分原因是需要丢弃具有不可纠正错误的条形码。其缺点包括需要预先设定基因组和探针、难以探测较短的转录本、较长的成像时间、对大面积组织的可扩展性有限、细胞分割中可能存在挑战以及需要处理 TB 的图像。
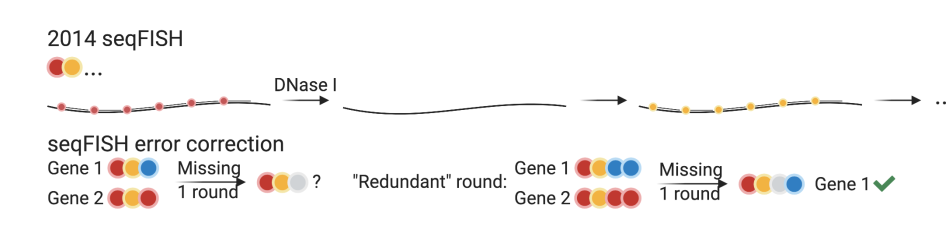
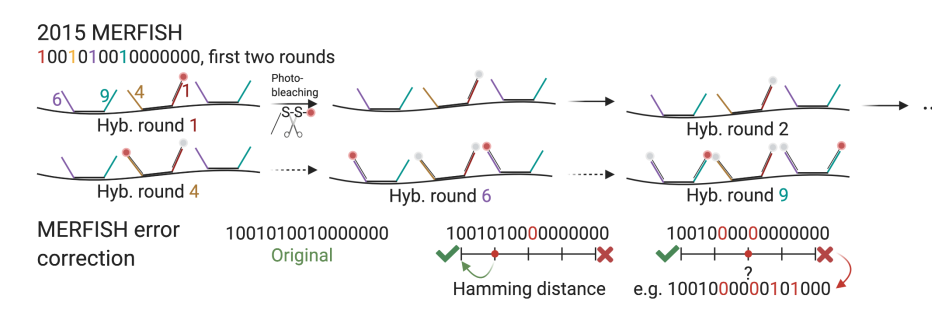
图5 seqFISH 及 MERFISH 原理示意图
靶向原位测序技术
ISS 方法通常通过基于连接(SBL)或者基于合成(SBS) 的原位测序, 确定靶向基因序列或非靶向的 cDNA 短片段,最终获得空间转录组信息。
根据测序链接策略的不同(SOLiD,cPAL和SEDAL),基于 SBL 的原位测序技术方法的代表性技术分别为 FISSEQ,ISS,STARmap。其中 ISS 方法最早于 2013 年出现,该方法使用挂锁探针来靶向已知基因。在完整的组织切片内,mRNA 首先被逆转录为 cDNA,挂锁探针可以结合到其上。挂锁探针是一种单链 DNA 分子,含有与目标 cDNA 互补的区域。虽然带有挂锁探针的 ISS 有利于亚细胞分辨率,但由于测序读出长度短和 RCP 的大小导致目标数量有限。ISS 技术在出现后被反复使用并被 CARTANA 商业化,该公司最近被 10X Genomics 收购。
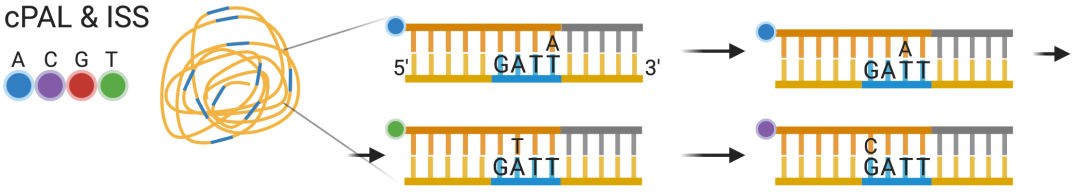
图6 cPAL&ISS 原理示意图
ISS 的优势包括单细胞分辨率和亚细胞转录本定位,缺点主要在于检测效率较低,早期最高分析基因数目为 39 个,目前最高报道为 222 个。
空间原位阵列捕获
转录本的空间位置信息可以通过在原位阵列上捕获的组织切片中的转录本来获取,目前已有多种技术策略被开发报道, 如空间转录组技术(Spatial Transcriptomics,ST)和 Visium 技术,通过将包含空间位置信息的条形码、UMI 标签、poly-dT 的探针固定在商用微阵列载玻片上,来捕获包含 poly(A) 尾巴的转录本,最终获取全转录组水平的基因表达及位置信息。在 Slide-Seq 中,探针通过微液滴将混合组合标签、UMI 标签和 poly-dT 置于单层载玻片上,而在 HDST 中,探针被置于载玻片上蚀刻的孔中。在 DBiT-seq 中,则通过微流体通道生成空间阵列,微通道用于在一个方向上提供一种类型的标签,然后在垂直方向上提供另一种类型的标签,形成正交连接通道,以便可以通过的成对组合标签识别每个斑点位置 。
基于空间原位阵列捕获的技术已经应用于面积较大的组织,并且它们的使用正在快速增加,空间分辨率也在逐步提高。ST 的光斑直径为 100 µm, 间距为 200 µm(图2 G)。Visium 是在 10X Genomics 收购 ST 后发布的 ST 的改进版本,其斑点呈直径为 55 µm 的六边形阵列。Slide-seq 中的磁珠直径为 10 µm,HDST 中的磁珠直径为 2 µm。Slide-seq 和 HDST 使用的微液滴大小小于细胞,但是它们仍然不能提供细胞水平的分辨率,因为一个微液滴可以跨越两个或更多细胞。DBiT-seq 的分辨率取决于通道宽度(50、25 或 10 µm)。在 2021 年出现了亚微米级别分辨率的空间新技术,其中之一是华大基因开发的 Stereo-seq,据报道,其光斑直径为 220 nm,而光斑之间的距离为 500 或 715 nm;另外一个是 Seq-Scope,空间位置标签之间的距离为 0.5 µm;最近的近亚微米技术是 PIXEL-seq,分辨率小于 1µm。
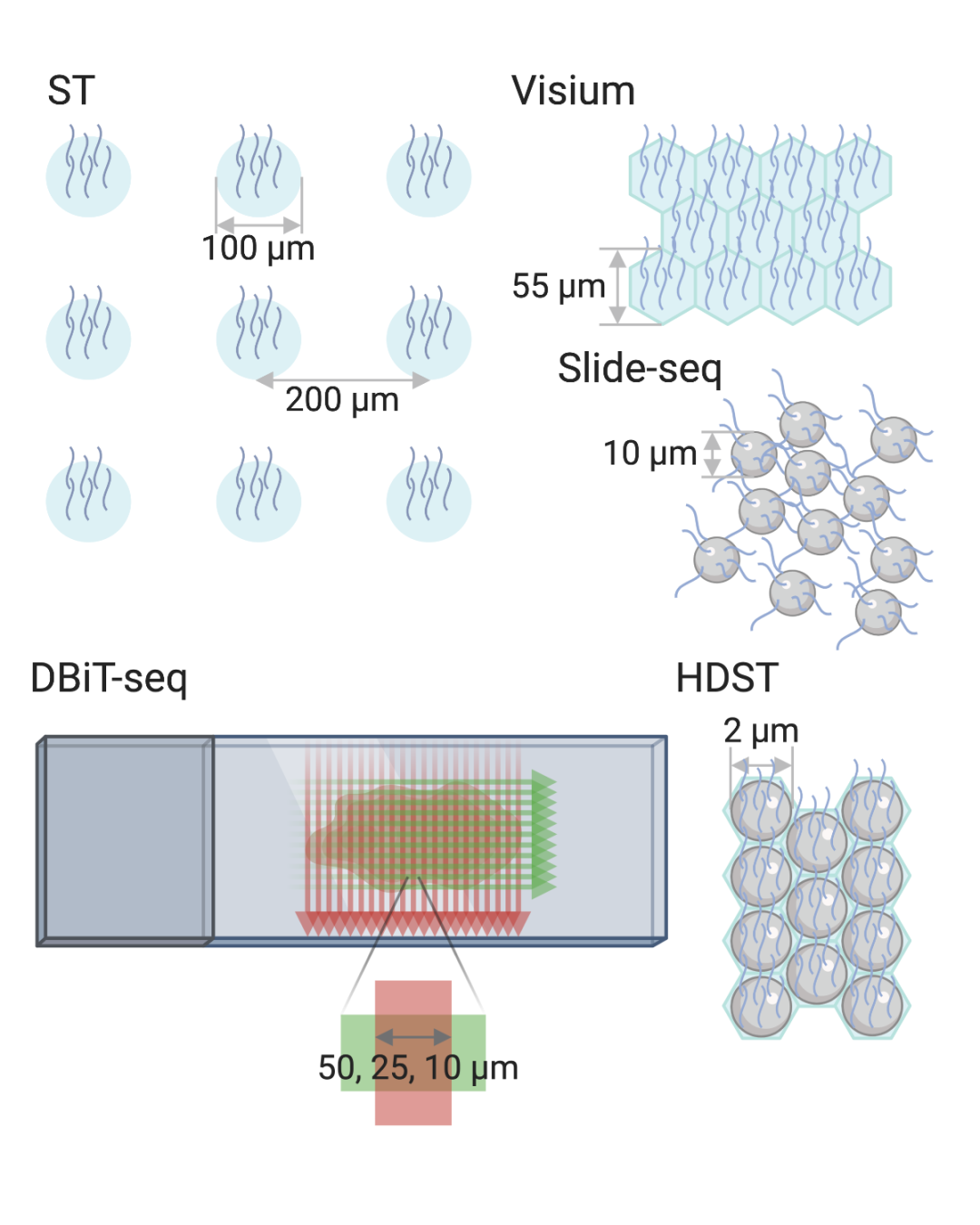
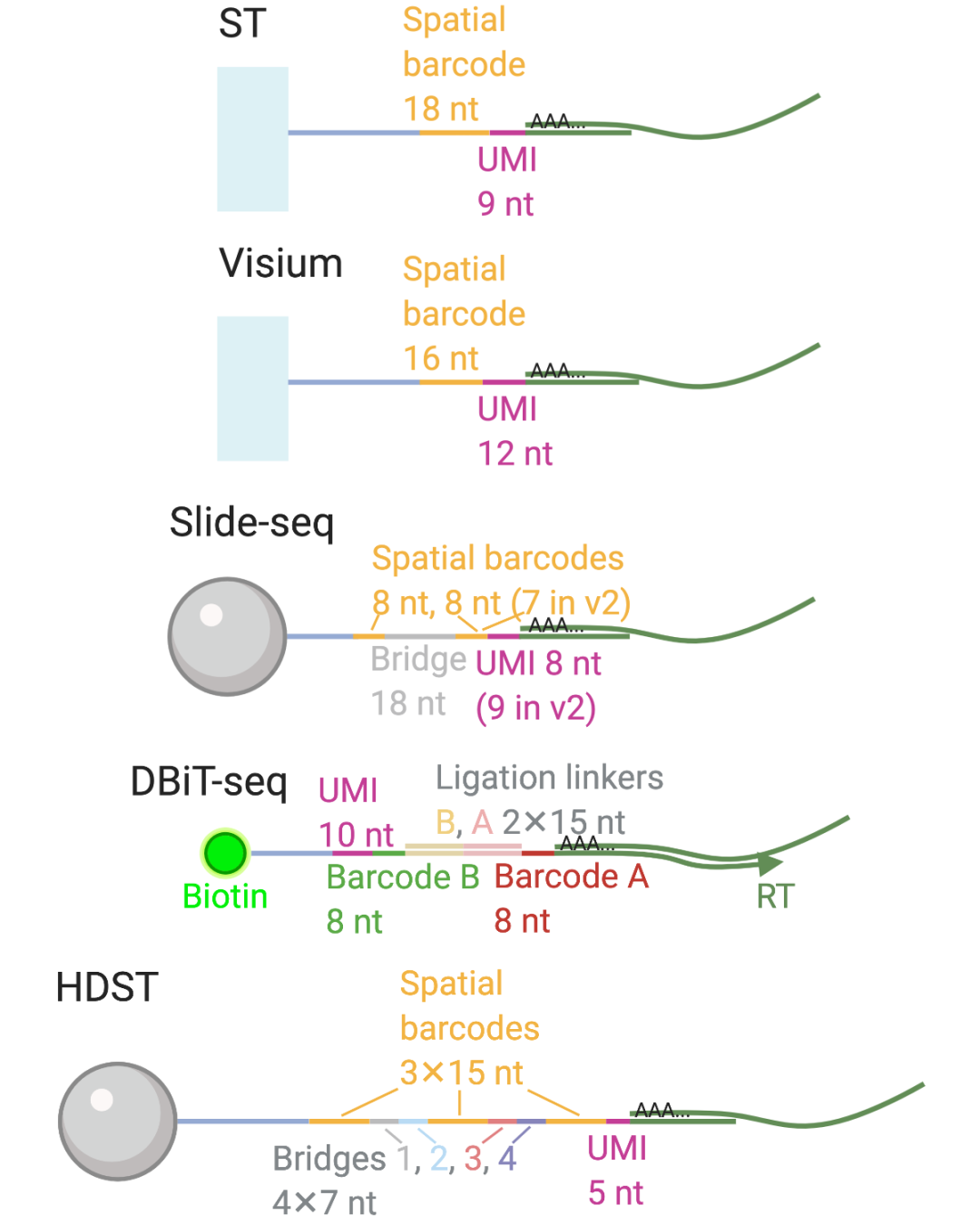
图7 几种空间原位阵列捕获技术原理及序列标签情况
不依赖成像的技术
除了前面提及的依赖于成像的空间转录组技术外,一些无需成像即可计算重建空间基因表达模式所需信息的技术也被开发了出来,其中之一就是是 DNA 显微技术,它记录 cDNA 之间的接近度,该信息可用于重建转录本之间的相对位置。另外,也出现了一些技术(如 pcRNAseq),在细胞水平上,可以通过多重分析来重建稀有类型细胞基因的空间表达情况,然后基于来自常见细胞类型的细胞基因表达将其映射到空间参考中的位置,这些细胞的类型与附着于稀有细胞类型的细胞相关。此外,“空间转录组学”这一术语的变体,也用于描述将转录本定位到细胞器的技术(例如 APEX-seq),尽管该技术没有记录任何空间坐标。
空间多组学
此外,发展的另一个方向是空间多组学。带有寡聚核酸标签的抗体可用于检测感兴趣的蛋白质,并且可以使用基于 smFISH 的方法检测表示蛋白质种类的寡核苷酸。此类抗体组合已与 ST 的变体相结合,如 SM-Omics ,GeoMX DSP 和 MERFISH 。
2.2 数据分析
高通量空间转录组学数据的处理和分析需要新颖的方法和工具支撑,尤其是在预处理、scRNA-seq 数据的空间重构、基于微阵列数据的细胞类型去卷积、空间高变基因的识别、细胞间相互作用的推导等。
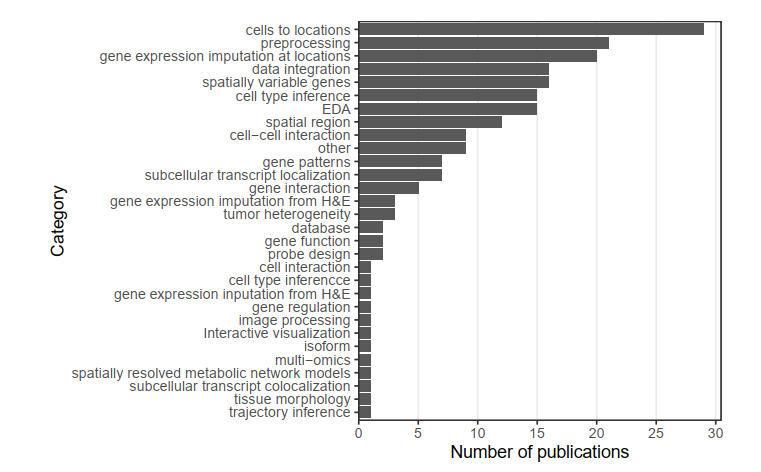
图8 空间转录组数据分析内容统计情况
数据预处理
“预处理”是指从原始数据中提取信息,以便可以应用常见的方法进行分析。“原始数据”可以表示任何形式的数据,即使以某种方式进行了处理,也仍然需要提取信息以应用常见的分析任务,例如 PCA、聚类和差异分析等。使用 NGS 的基于空间微阵列捕获技术的预处理类似于 scRNA-seq 的预处理。对于基于 smFISH 和 ISS 的数据,原始数据是包括荧光斑点的图像,必须对其进行处理,以进行转录本识别, 并进行细胞分割。
scRNA-seq 数据的空间重构
最近,scRNA-seq 技术的进步启发了利用高分辨率转录组定量分析与空间转录组学数据的互补性质的新方法。对于未进行转录组测序的 smFISH 和 ISS 数据,可通过 scRNA-seq 数据估算空间数据中未分析的基因的表达模式,方法是将解离的 scRNA-seq 细胞映射到空间参考区域,或直接使用来自 scRNA-seq 的表达谱来估算空间中的基因表达。也可以从 scRNA-seq 推断空间中的基因表达,而无需明确将 scRNA-seq 细胞映射到位置,一种常见的方法是将空间和 scRNA-seq 数据投影到共享的低维且无批次的潜在空间中,然后通过将空间细胞投影到潜在空间中来估计基因表达。这种方法的示例包括 Seurat3 和 gimVI。当某些多组学数据的空间技术不可用时,也可以使用这些方法将空间位置信息添加到单细胞多组学数据中。
细胞类型推断
在没有达到单细胞分辨率的空间数据(例如由 ST 和 Visium 衍生的空间数据)中,通过整合 scRNA-seq 数据,可以推断每个 spot 的细胞类型组成。一种常见的策略是将在 spot 处观察到的基因表达作为来自 scRNA-seq 的每种细胞类型的平均基因表达的加权总和进行显式建模,然后可以基于负二项式或泊松分布统计模型对基因计数进行建模,最后可以根据模型的参数估算每个点中的细胞类型比例,采用该策略的软件包括 stereoscope 和 RCTD。
空间高变基因
空间高变基因是其表达与空间位置相关的基因。通常使用两种方法:高斯过程回归(GPR)和拉普拉斯评分(Laplacian)。前者将归一化的基因表达的速率参数建模为 GPR,并找出该模型是否更好地描述了带有空间项的数据而不是没有空间项。为了加快计算速度,可以在应用 GPR 方法之前将细胞或斑点与自组织图进行汇总。后一种方法确定了其表达能更好地反映空间邻域图结构的基因。细胞的位置也可以被建模为一个以基因表达为标记的空间点过程,可以将空间可变的基因鉴定为与位置相关的标记。基于 GPR 的方法从理论上近似基因的 p 值,而其他方法则使用置换测试,这使得它们的可扩展性较差。但是,通常用于基于 GPR 方法的高斯核不能解释在组织中观察到的各向异性。
细胞通讯
和单细胞转录组一样,空间转录组也可以进行细胞间通讯分析,区别在于空间位置信息可以同时用于识别潜在的细胞间相互作用。通常通过了解配体受体(LR)对并测试 LR 对是否更有可能在邻近细胞或 spot 中表达来完成,还可以对目的基因的表达进行建模,包括细胞-细胞共定位的术语。
探索性分析生态系统
鉴于 scRNA-seq 与空间数据的相关性,已有一些流行的 scRNA-seq 探索性数据分析(EDA)生态系统,例如 Seurat ,SCANPY(Squidpy)和SingleCellExperiment(SpatialExperiment)等,通过对软件和功能的更新,实现了空间位置的基因表达和细胞/spot 位点的可视化。另外具有图片精美、文档完善的空间数据的 EDA 软件包也已经被开发出来,例如 Giotto、STUtility 和SPATA 等。Seurat、Giotto 和 SPATA 还封装了空间高变基因识别的一些基本方法。此外,Giotto 开发或者封装一些方法来识别 ST 和 Visium spot 位点中的细胞类型富集解析、基因共表达识别、基因表达与细胞类型共定位之间的关联以及识别空间区域。
其他分析
除以上所述之外,还有很多其他类型的分析可用于空间转录组学数据,包括基因模式的识别,空间区域的转录组定义, 基因间的相互作用推断,转录本的亚细胞定位以及基于H&E图像的基因表达估算等。
3. 领域趋势
现有技术在质量与数量之间的折中意味着目前尚无单一的“最佳”解决方案,并且由于操作上的困难导致许多技术从未应用到发明机构之外的其他机构。目前,Visium、ST、Tomo-seq、LCM 和 ISS 技术使用最为广泛。就分析的组织而言,当前已广泛用于研究人体组织、肿瘤(尤其是乳腺肿瘤和鳞状细胞癌)以及不一定具有定型结构的病理组织(图9)。在 SARS-CoV-2 大流行期间,GeoMX DSP 已被用于在 COVID 死亡患者的病肺上进行空间转录组研究。
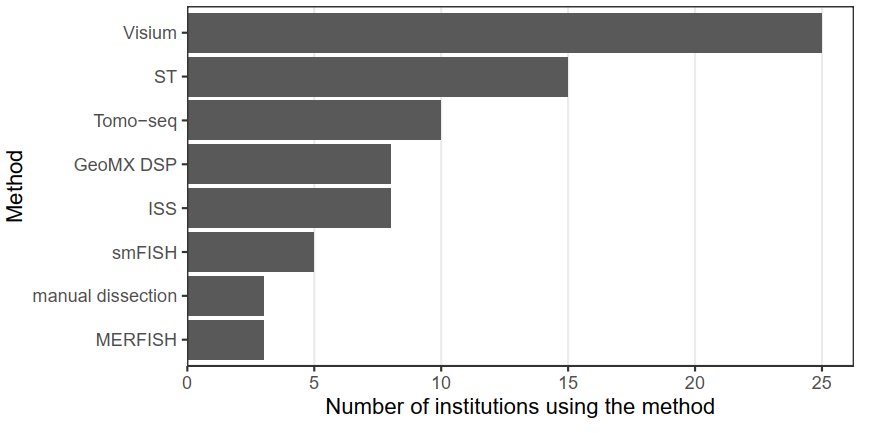
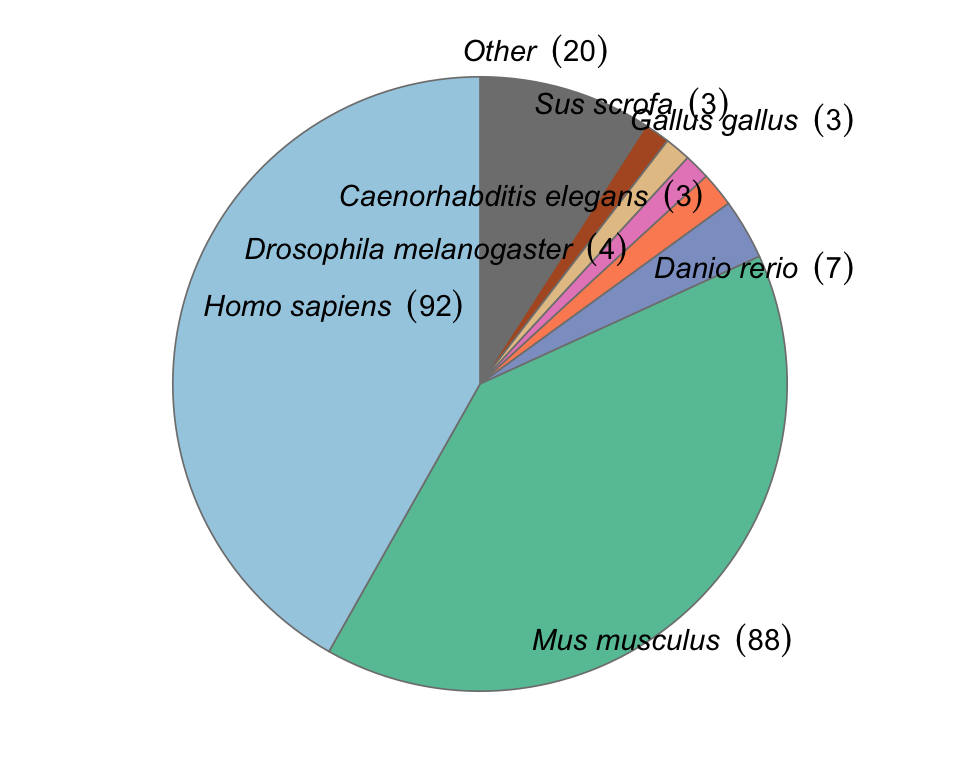
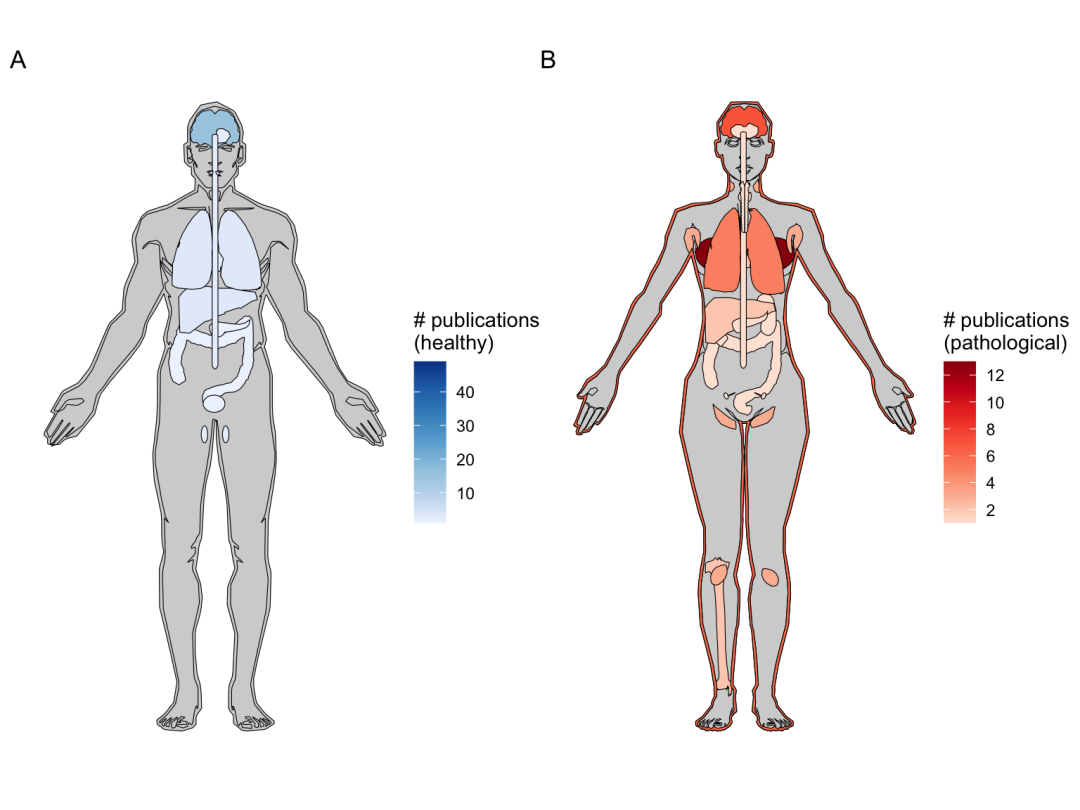
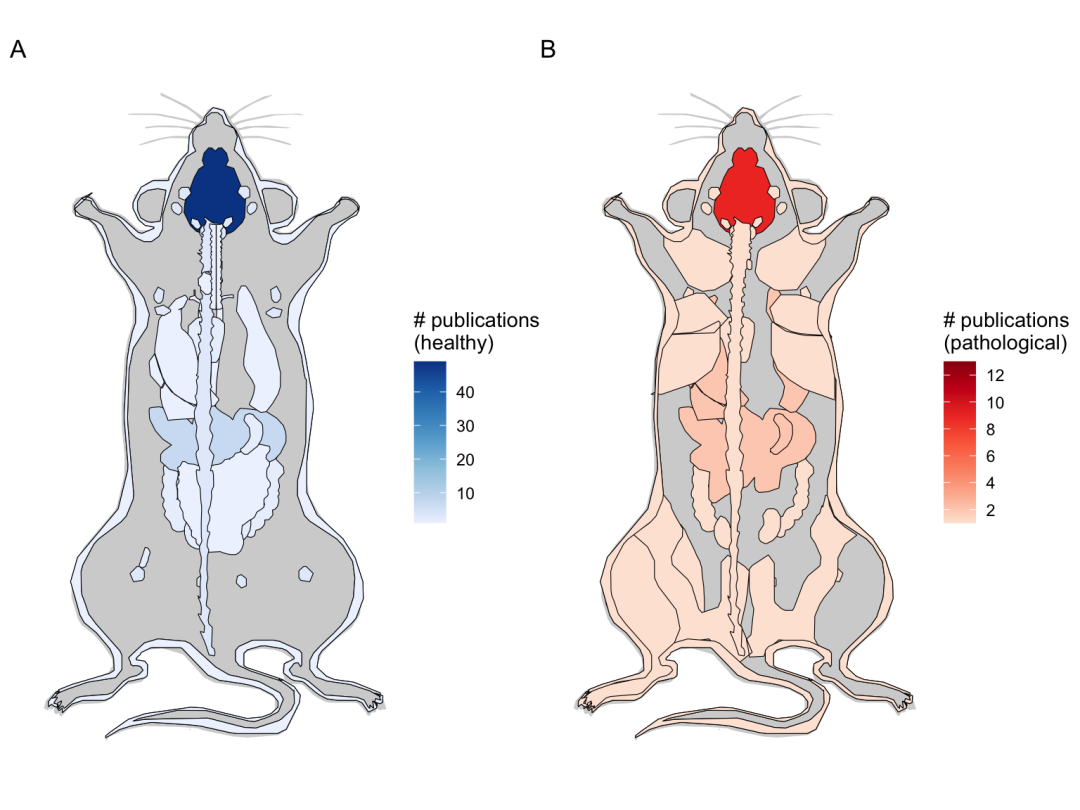
图9 空间转录组研究统计情况
除了 LCM,当前时代的绝大多数研究都是在人类或小鼠身上进行的(图9),而大脑是研究最多的器官(图9)。尤其是美国脑计划启动的 BICCN 项目,该项目将整合不同类型细胞的分子、解剖学和生理学特性,创建小鼠脑细胞 3D 参考图谱,同时也将基于人类和非人类灵长类脑组织样本,绘制脑细胞参考图谱 ,其涉及空间转录组学技术主要包括 MERFISH 和 seqFISH。尽管此项目中使用的 smFISH 技术原则上可以扩展到许多基因,但实际上,它们在大多数情况下研究的基因(图10)和细胞(图10)数目有限。
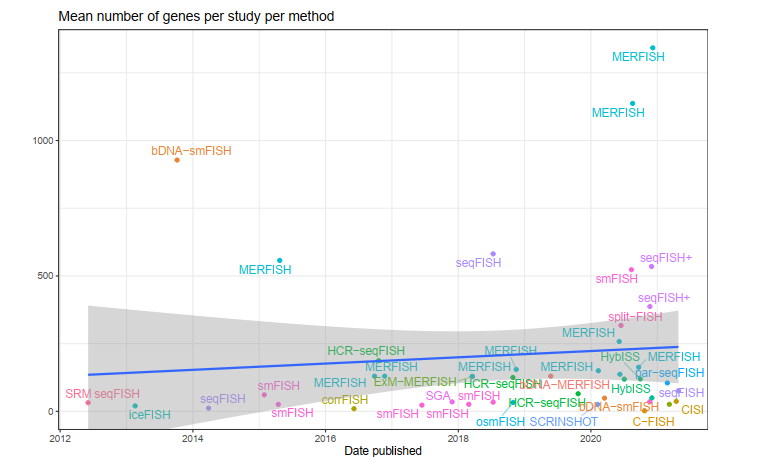
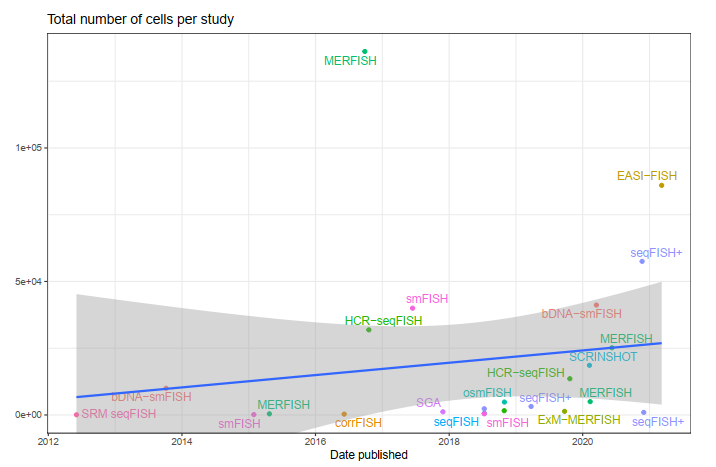
图10 MERFISH 和 seqFISH 研究统计情况
文档中数据分析部分提到的所有软件包都是开源的,并且使用 R、Python 和 Julia 等语言编写。在主要涉及新数据和数据分析包的研究中,下游分析主要使用诸如 R、Python 和 C++ 之类的开源编程语言(图11 )。尽管 MATLAB 仍然很流行,但随着 R 和 Python 的流行,它的使用似乎在下降。R 在下游分析和 EDA 中更受欢迎,而 Python 和 C ++ 在包开发中更受欢迎(图11 ),反映了这些语言及其周围文化的相对优势。大多数软件包都不托管在标准存储库中,例如CRAN,Bioconductor,pip 和 conda。
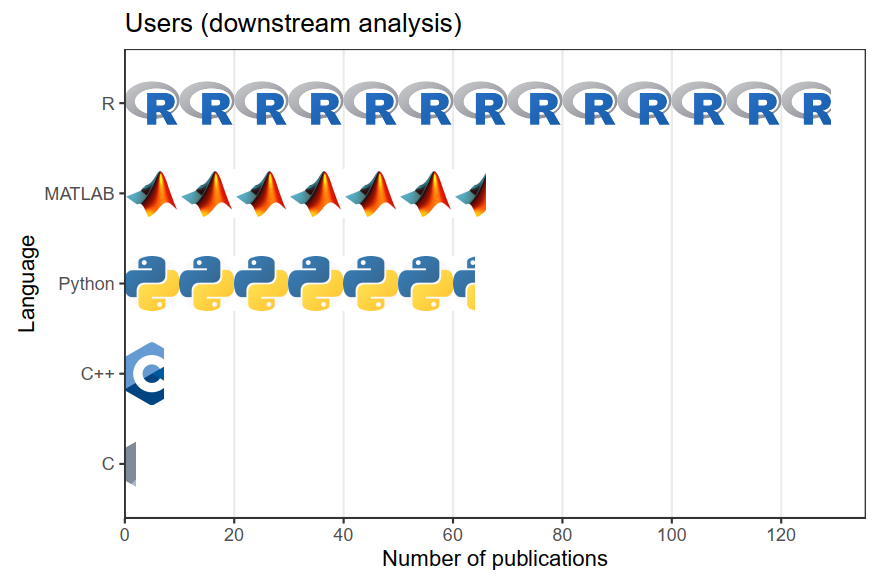
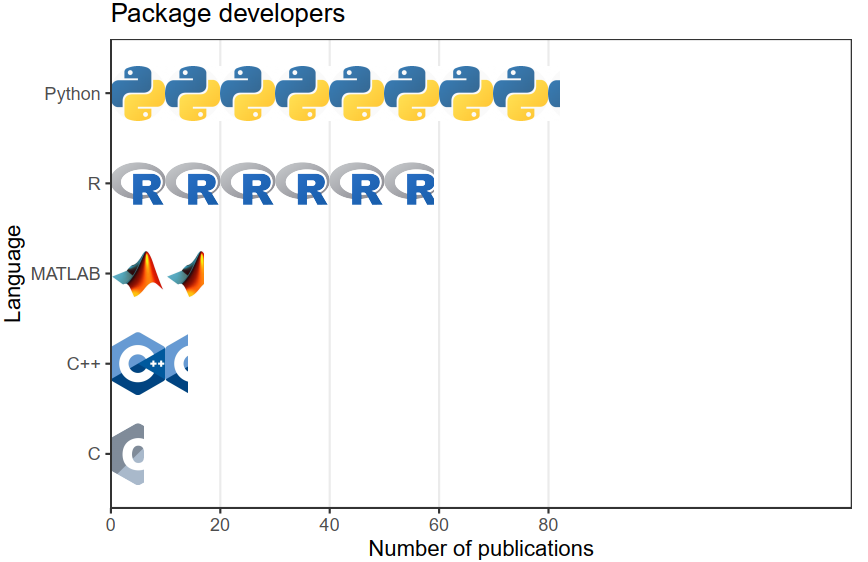
图11 空间转录组数据分析软件开发语言统计情况
4. 未来展望
尽管前传时代的空间技术被迅速弃用,但其思想和方法是当今时代空间转录组学发展的基础。在过去的 5 年中,随着众多新技术的出现和 10x 公司 Visium 技术解决方案的商业化普及,该领域得到了快速的发展(图12)。对各种组织的适用性以及商业工具包和核心设施的可用性,减少了安装仪器和培训人员的工作和成本,从而推动了这些技术的普及。
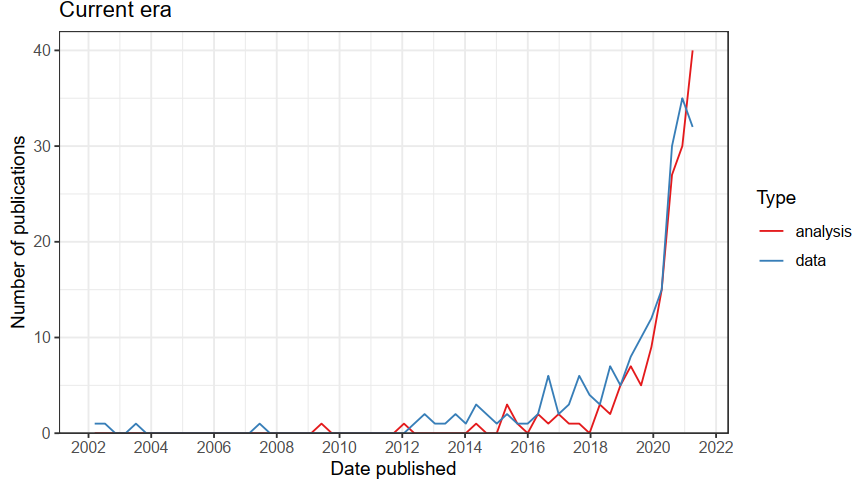
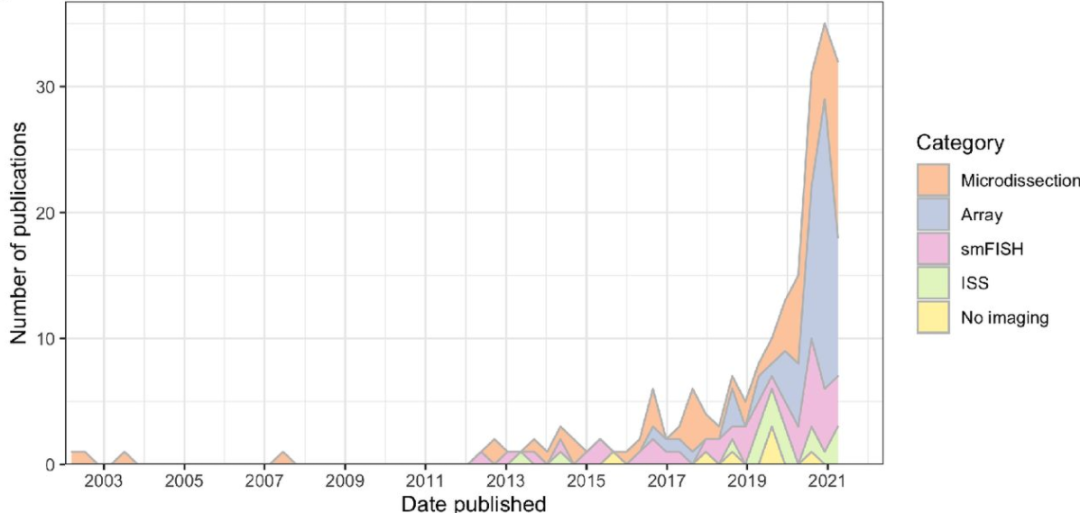
图12 空间转录组数据发展水平统计
尽管如此,当前空间转录组学仍然面临许多挑战:
第一,实验技术有待改进。我们可以做更多的事情来改善实验技术。例如 组织切片技术的发展、 smFISH 和 ISS 技术的实验技术优化、高效率的计算工具实现真正的 3D 空间转录组或者空间时序转录组等。
第二,数据库的建立。尽管当前空间转录组的数据在快速地增长,但尚未有对应的数据库来对这些数据进行汇总集成。
第三,研究物种及组织的多样化。在当前时代,除 LCM 之外,研究的器官主要集中于人类和小鼠的大脑,而其他器官(例如肝脏)的空间转录组学研究相对滞后。对除人类和小鼠以外的其他生物以及除大脑以外的其他器官的技术现代化,将为开发有用的空间转录组学地图集提供广阔的前景。
第四,集成度高、交互性强、易扩展、文档信息完善的开源工作流程的开发。该类工具的开发将大大简化不同类型空间转录组学数据的整合和分析。这类工具应该像 SeuratWrappers 一样,它为数据集成和 Seurat 包中未实现的 RNA 速率分析提供 Seurat 接口,或者像 caret 和 tidymodels 一样,它们为多种机器学习方法提供了统一的用户界面。一般可以通过诸如 Bioconductor 所使用的准则之类来实现,鼓励开发人员重用 Bioconductor 中的现有数据结构和方法,而不是重新发明轮子。当前,对于 EDA 以外的任务,用户仍然经常需要学习新的语法,转换对象类型,甚至学习新的编程语言以使用某些数据分析,空间转录组学仍然面临许多挑战。
5. 参考资料
Moses L, Pachter L. Museum of Spatial Transcriptomics[J]. 2021.