






继上期《喜报!喜报!这两项软著授权,科研人的福音来啦~》之后,近期青岛欧易基因组学在证书授权上又迎来好消息啦~此项授权软件为SNP一致性比较工具软件,这标志着青岛欧易在基因组技术的专业性更进一步,同时意味着青岛欧易将能更好的为广大科研工作者服务~

SNP一致性比较工具软件
单核苷酸多态性(single nucleotide polymorphism,简称SNP)主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性,它在基因组中广泛存在。SNP是一种二态的标记,由单个碱基的转换或颠换所引起,也可由碱基的插入或缺失所致。SNP既可能在基因序列内,也可能在基因以外的非编码序列上。用于存储基因分型变异位点的标准格式是Variant Call Format(简称VCF)。
现在测序技术飞速发展,新的测序平台不断涌出,评价新测序平台检测出SNP的稳定性、准确性等指标极为重要。同一样品通过待评价的新测序平台和“金标准”的测序平台分别检测出的SNP同时储存在VCF格式的文件中,本软件可以分别比较两者间纯合位点和杂合位点的一致性,从而间接性评估待评价的新测序平台的可靠性,为下一步是否可替换为价格更低的新测序平台提供依据。
使用说明
01 核心功能
本软件可以计算重复样品、实验对照样品及任何两个样品之间SNP的一致位点数和不一致位点数,并对所有位点进行分类统计,从而确定优化后的实验流程、分析流程以及测序平台等是否有较好的效果。
02 基本说明
软件需要有格式正确的输入文件方可正常执行。
其使用样例如下:
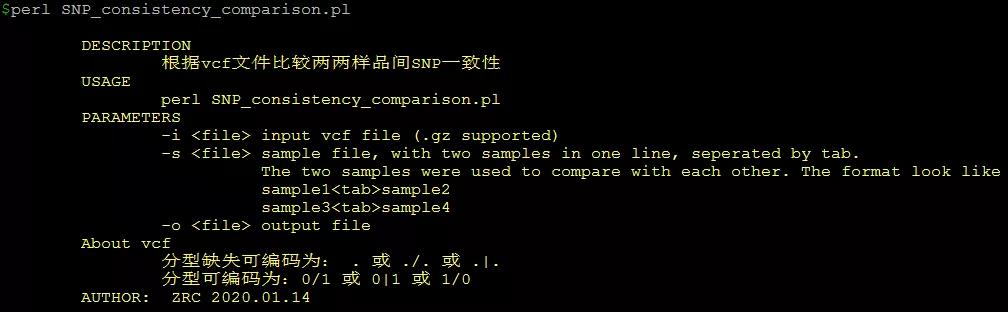
必要参数:
-i 输入基因分型VCF格式数据,支持gzip压缩格式
-s 两两样品比较分组列表,使用tab分割
-o 输出信息统计表
03 使用示例
perl SNP_consistency_comparison.pl -i AN03.imp_hap.vcf.gz -s sample_compare.list -o result.txt
其中,-i AN03.imp_hap.vcf.gz 为基因分型VCF格式数据(输入文件);-s sample_compare.list为需要比较的两两样品名称列表文件(输入文件);-o result.txt 为输出统计表文件
软件输出结果展示
信息统计表文件(-o参数),如下图。

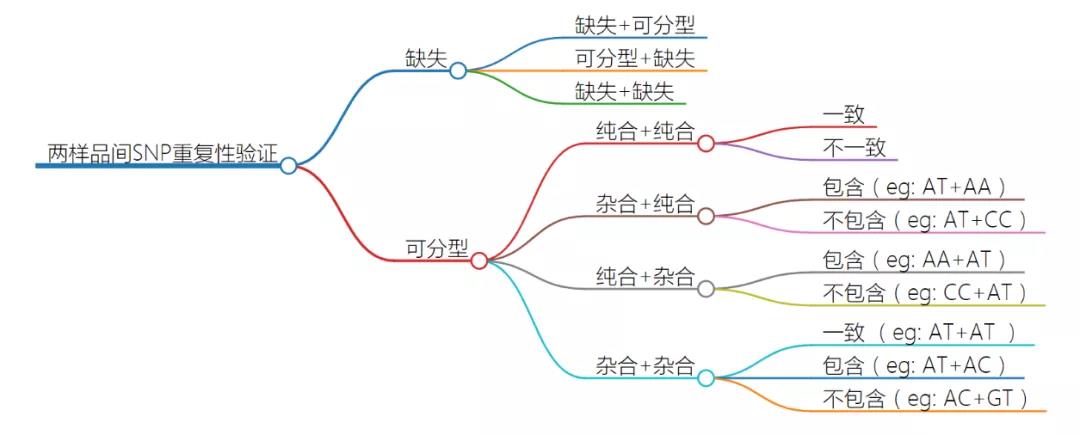
结果格式说明:
sample1列:第一个样品名称;
sample2列:第二个样品名称;
miss_genotype列:第一个样品没有分型且第二个样品有分型的位点数,如“.”与“AA”;
genotype_miss列:第一个样品有分型且第二个样品没有分型的位点数,如“AA” 与“.”;
miss_miss列:两个样品都没有分型的位点数,如“.” 与“.”;
homo_homo_identical列:两个样品分型均为纯合且一致的位点数,如“AA”与“AA”;
homo_homo_discordance列:两个样品分型均为纯合且不一致的位点数,如“AA”与“TT”;
hete_homo_include列:第一个样品为杂合,第二个样品为纯合且纯合碱基包含于杂合中的位点数,如“AT”与“AA”;
hete_homo_notin列:第一个样品为杂合,第二个样品为纯合且纯合碱基不包含于杂合中的位点数,如“AT”与“CC”;
homo_hete_include列:第一个样品为纯合,第二个样品为杂合且纯合碱基包含于杂合中的位点数,如“AA”与“AT”;
homo_hete_notin列:第一个样品为纯合,第二个样品为杂合且纯合碱基不包含于杂合中的位点数,如“CC”与“AT”;
hete_hete_identical列:两个样品均为杂合且一致的位点数,如“AT”与“AT”;
hete_hete_include列:两个样品均为杂合且只有一个碱基一致的位点数,如“AC”与“AT”;
hete_hete_notin列:两个样品均为杂合且两个碱基均不一致的位点数,如“AC”与“GT”;
下图可辅助理解,“+”前后分别为第一个样品和第二个样品
青岛欧易
青岛欧易生物科技有限公司多年来,立足于生命科学,聚焦基因组学、分子育种、微生物等领域。公司现拥有2b-RAD、MethylRAD、Super-GBS、LcWGS、2bRAD-M微生物五项特色技术,经过多年努力,人才队伍更加壮大、团队管理经验更加丰富、技术服务工作更加精准高效,积极参与并推动科技服务产业发展,致力于成为受人尊敬的生物科技公司。
END
青岛欧易 撰文
本文系欧易生物原创
转载请注明文本转自欧易生物