






随着三代测序技术的发展,PacBio SMRT测序技术应用场景越来越广泛。与二代测序方法相比,采用PacBio SMRT长读长测序技术的三代宏基因组可以减少部分拼接错误,提高基因组组装注释的准确性和微生物群落鉴定的分辨率。上海健康医学院和上海市浦东新区周浦医院李延飞老师团队在Cell Death and Disease期刊发表了人肠道三代宏基因组测序的相关成果,鉴定了人肠道菌群的一个新菌种Enterococcus tongjius,值得一提的是,欧易生物张建明被列入了本篇文章的署名作者。

基本信息
材料:2个人粪便样本
期刊:Cell Death and Disease
发表时间:2021年6月2日
方法:欧易生物三代宏基因组测序(PacBio SMRT测序)
研究背景
人体大多数微生物存在于肠道中,它们在合成机体必须的维生素和营养素、保护宿主免受病原体的入侵及调节免疫宿主反应方面发挥重要作用。肠道菌群与宿主及外部环境保持动态平衡。宏基因组是微生物组样本中存在的所有遗传物质的集合,尽管已有150,000 个人类微生物基因组,但仍有很多肠道微生物群未确定。Edoardo et al.报道了近5000个种级别的基因组,但其中77% 未包含在公共数据库中。微生物鉴定的难点在于很多微生物不可培养,而高通量测序弥补了这些不足,因此高通量测序在菌群多样性分析和菌群鉴定等的应用也越来越广泛。
研究结果
1、 2个样本的肠道菌群基因组特征
对1名10月龄婴儿和1名34岁成年人的粪便样本DNA进行了PacBio宏基因组测序,分别获得的数据量为91.7 GB和81.7 GB。在儿童样本中,获得了43 contigs (≥0.5 Mb)和248 contigs(≥0.1 Mb)。在成人样本中,获得了33 contigs (≥0.5 Mb)和237 contigs(≥0.1 Mb)(图1b)。此外,contig长度与CDSs数显著相关(图1c)。
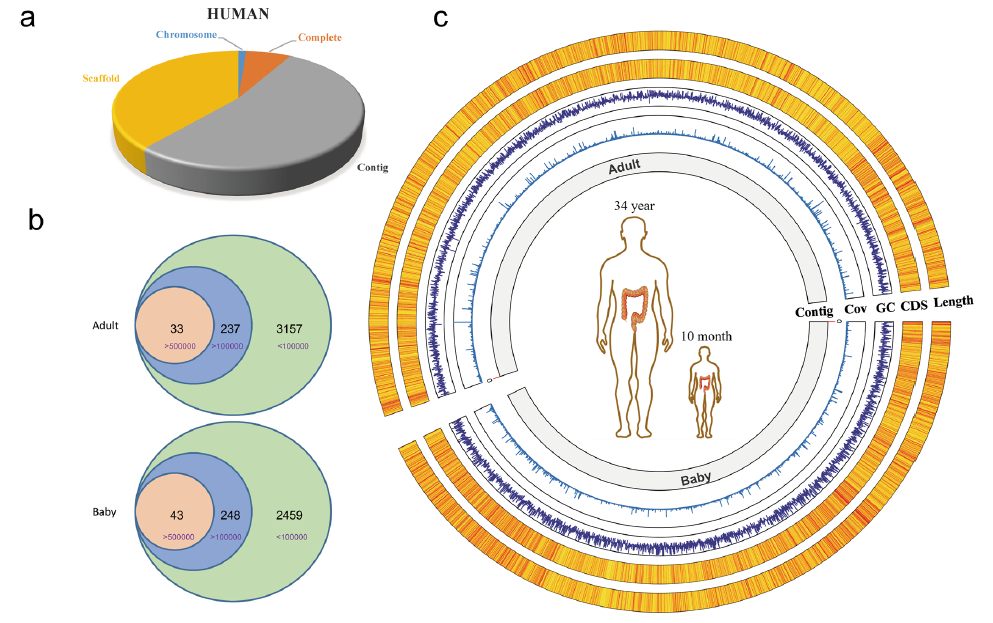
图1 两个样本肠道菌群基因组特征。a:NCBI 数据库中人类细菌序列数据,包括chromosome, complete, scaffold和 contig;b 两个样本中获得的不同长度的contigs数量;c 两个样品的基因组特征。
2、2个样本中长度大于0.5 Mb的contigs特征
大多数细菌基因组大于 0.5 Mb,因此进一步分析了长度大于0.5 Mb的contigs。10 个contigs是circular(图 2a),12 个contigs达到99% complete(图 2b),9个contigs是circular且达到99% complete(图2c)。对这9个contigs做进一步鉴定分析,和全长16S rRNA 序列进行比对分析,其中4个contigs可以和 NCBI数据库中的序列完全匹配(图2d和2e),5个contigs不完全匹配,表明可能属于新的种或亚种。同时系统进化树分析成功的确定了它们的分类学位置(图2e)。
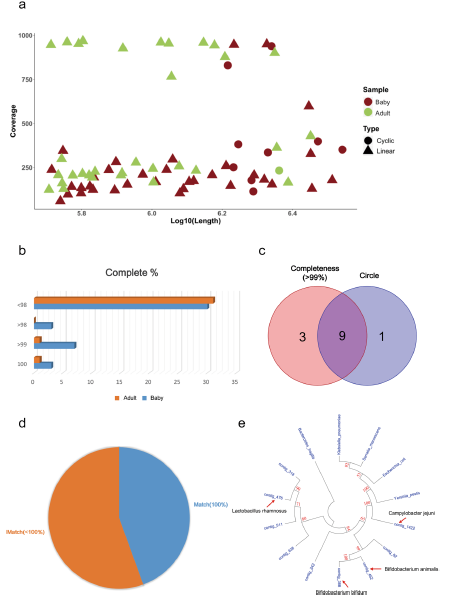
图2 两个样本中长度大于0.5 Mb的contigs特征。a-c:contigs的长度和覆盖度;d-e:9个contigs的blast比对和系统进化树分析。
3、5个不完全匹配contigs的基因组特征
对这5个contigs不完全匹配做进一步分析,其长度分布在1.5-3.5 Mb,尽管基因组长度差异很大,但平均长度相似(图3a)。contigs的特殊结构分析表明:rRNA数和tRNA数相关(图 3c)。每个contigs具有所有类型的重复序列,simple repeats是主要的重复类型(图 3d)。有意思的是,发现所有五种细菌都表现出链霉素耐药性(图 3e),表明抗生素的广泛使用可能已经不可逆转地改变了人肠道菌群。为了确定它们属于哪个分类,和 NCBI数据库中全长 16S rRNA进行比对,其中contig_82,contig_ 242、contig_318 和 contig_511的序列相似度在99%以上,而 contig_638 的序列相似度小于 97%(图 3f)。
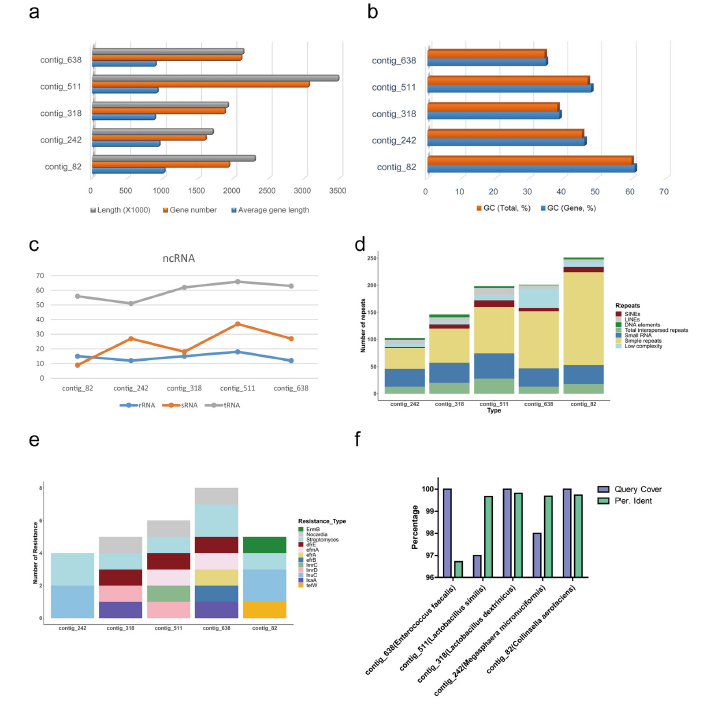
图3. 5个不完全匹配contigs的基因组特征。a-b:non-match genomes的长度和GC含量;c-d:non-match genomes的ncRNA和重复序列分析;e:non-match genomes的耐药性分析;f:non-match genomes的全长16S rRNA比对分析。
4、4个high identity non-matched基因组的进化分析
对4个high identity non-matched基因组(contig_82,contig_ 242、contig_318和contig_511)做进一步进化树分析。Contig_82属于Collinsella erofaciens并且可能是一个新的亚种(图 4a)。Contig_242是Megasphaera micronuciforrnis的一个亚种,contig_318是Lactobacillus dextrinicus的一个亚种,contig_511是Lactobacillus smilis的一个亚种(图4b-d)。全长比对分析发现:它们之间的差异是小,contig_318中只有5个碱基的差异(图4e)。Megasphaera micronuciforrnis (contig_242) 是一种条件治病的病原体,因此也评估了健康人是否也携带其他条件性或常见病原体。选择了27 个病原体,包括E. coli, Staphylococcus aureus, Shigella flexneri和Helicobacter pylori,仅有S. flexneri鉴定到,其他致病菌未鉴定到(图4f)。
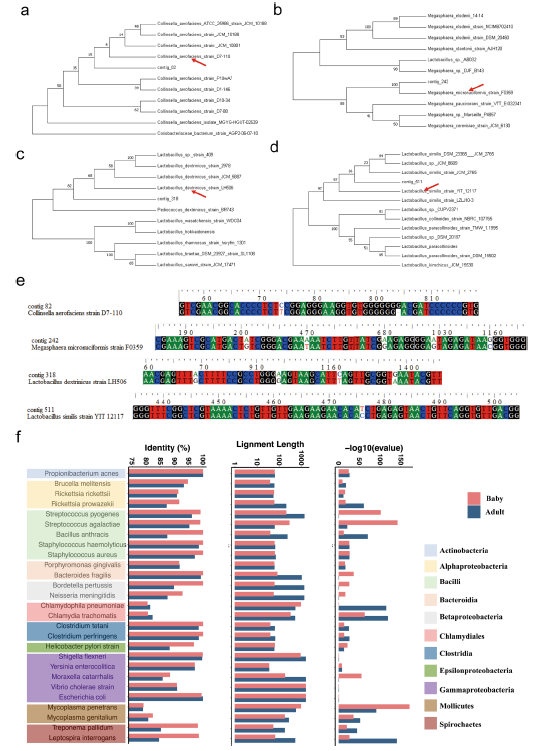
图4. 4个high identity non-matched基因组的进化分析。a-d:进化树分析;e:全长比对分析分析;e:non-match genomes的耐药性分析;f:致病菌分析。
5、新菌种Enterococcus tongjius的基因组特征
分析了contig_638,其中和其十分相似的Enterococcus faecalis仅显示8%的相似性(图5a)。通过NCBI blast和系统发育树分析发现contig_638属于Enterococcus(图 5b)。Enterococcus tongjius是一个全新的物种(图 5c),与其他Enterococcus共享867个核心基因,有 1162 个distinctive genes(图 5d)。基因组的功能分析发现:许多基因可能与碳水化合物和脂质代谢有关(图 5e),糖代谢的进一步分析表明,大多数基因与水解有关(图 5f)。Enterococcus tongjius基因组大小为2.1 M,GC含量为34.49%,与 其他Enterococcus一致(图 5g)。 Enterococcus tongjius有 2111 个基因,12 个 rRNA 和 63 个 tRNA(图 5g)。
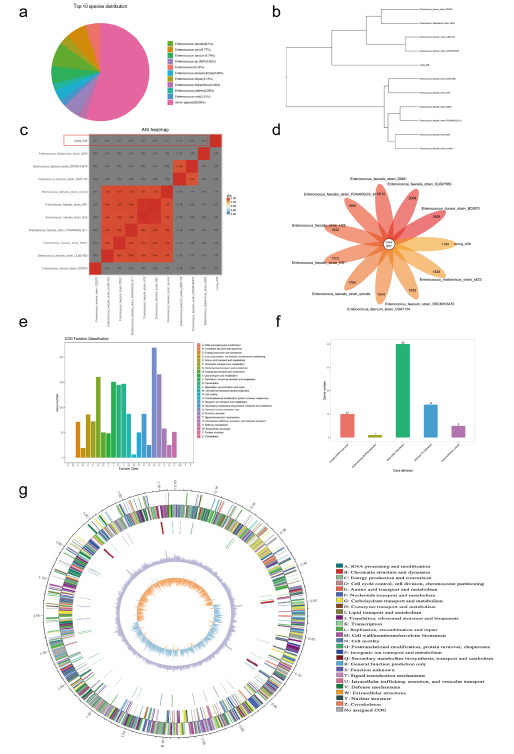
图5. 新菌种Enterococcus tongjius的基因组特征。a-d:新物种Enterococcus tongjius鉴定;e-f:基因组功能分析;g:Enterococcus tongjius的基因组Circos图。
参考文献
Li Y, Jin Y, Zhang J, et al. Recovery of human gut microbiota genomes with third-generation sequencing. Cell Death & Disease. 2021 Jun;12(6):569. DOI: 10.1038/s41419-021-03829-y.