






基因型填充的含义
基因型填充是一个根据参考面板(reference panel)中的单倍型和基因型预测和填充缺失基因型的过程。基因型填充是基于这样的假设,即两个个体,即使明显无关,在其基因组中共享来自遥远共同祖先的short panel。因此,可以通过包含大量标记的panel来推断一个样本中未观察到的基因型。大多数当代插补工具采用隐马尔可夫模型框架,从reference panel中估计单倍型推断基因型(r2显示出高度连锁不平衡)。
其实,简单来讲就是,根据样品中已知位点分型信息去reference panel(样品量足够大、品种足够多的包含各种组合可能的单倍型数据库)中匹配最相似的来填充样品中缺失基因型,其示意图如下:
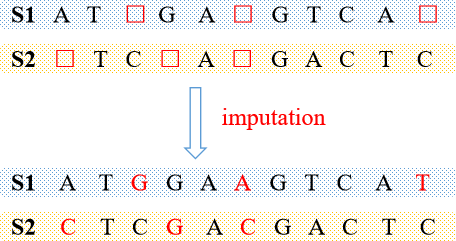
图片说明:S1和S2代表样品,口 表缺失基因型,A、T、C、G代表已知基因型
基因型填充的意义
目前高通量SNP检测方法主要包括全基因组重测序(WGS)、SNP芯片、简化基因组测序等。其中全基因组重测序理论上能检测基因组范围的所有变异位点,但是测序成本高昂;SNP芯片检测标记数量有限、只能检测已有突变,目前商业化的SNP芯片只包含部分物种;简化基因组测序技术相较于SNP芯片,检测位点更多且不受物种限制,但只能检测基因组2%左右的变异位点。
有的同学说了,现在测序这么便宜,测个重测序还测不起么。一个两个、十个二十个当然测得起,但是对于全基因组关联分析(GWAS)和基因组选择(GS)应用而言,测序样本量一般都是几千甚至几万。这样的话,即使是基因组较小的水稻,那也是一笔不小的费用。
那么有什么省钱的方法么?当然有了,如果将SNP芯片、简化基因组测序等检测变异位点较少的技术,利用reference panel填充的方式达到WGS检测的位点数目不就好了么。这样,既省了钱又能拿到百万级、千万级的变异位点。
基因型填充方法
小编目前知道的填充方法有两种。
一种是基于任何技术得到的变异位点Variant Call Format (VCF)文件,基于reference panel对VCF进行填充,最终填充到位点数与reference panel相同。
另一种是基于低深度重测序(LcWGS)的方法,即同WGS一样对样品构建文库,但是测序只测1X甚至更低。分析上则利用比对后的bam文件以及reference panel对样品直接分型,最终获得与reference panel相同的位点数。
有同学问了,这两种方式哪种更好呢?小编认为第二种更有优势,原因有以下几点:
VCF文件比bam信息含量更少;VCF只包含了突变位点,本次测序的群体中没有突变的位点被屏蔽或过滤掉,但这不意味着在其他群体中没有突变;
bam文件中存储的reads,在R1和R2之间存在一定的相位信息,这些相位信息是极其重要的;
LcWGS比其他技术得到的reads对基因组覆盖范围更广,这样与reference panel中有重叠的点会更多。
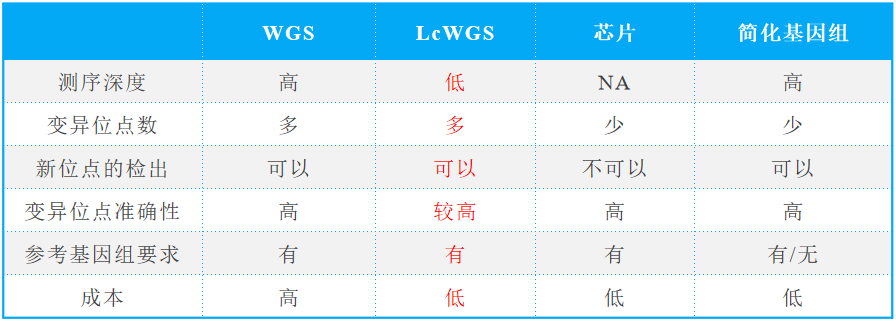
不同技术之间的比较
全基因组重测序、SNP芯片、简化基因组测序和低深度重测序技术比较如下:
reference pane
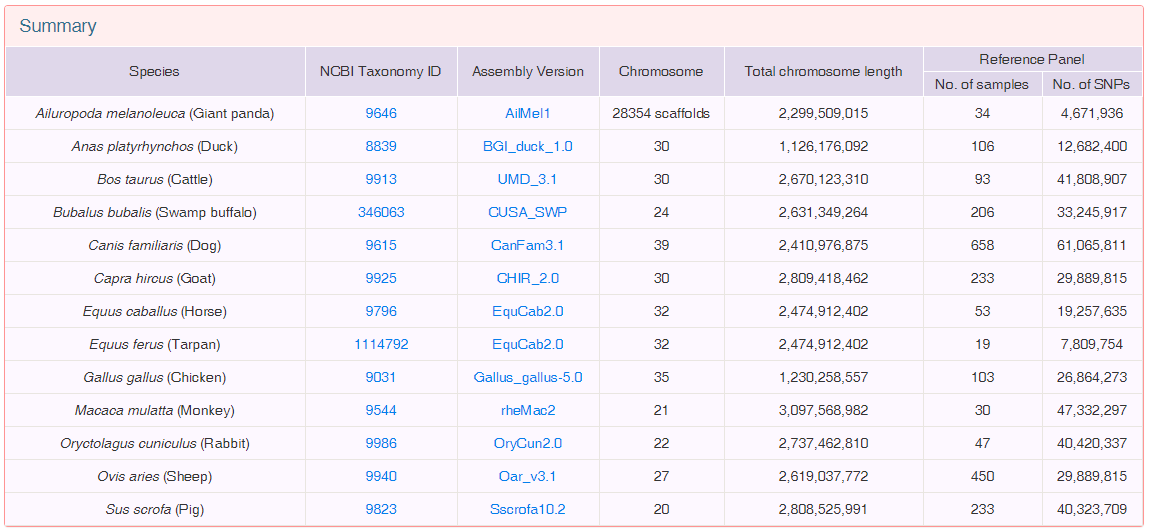
有同学说,你光说填充多好多好,没有panel怎么办呢?别急,华中农业大学构建了13种动物和12种植物的reference panel,并且数据库公开(感谢大佬),快去看看自己研究的物种在不在里边吧。现将网址附上:
动物 http://gong_lab.hzau.edu.cn/Animal_ImputeDB/#!/

植物 http://gong_lab.hzau.edu.cn/Plant_imputeDB/#!/

对于没有reference panel的物种,大家也不要急,有两种方法可供大家使用。一是可以参照华中农大文章的方法搜索已有的数据或自行测序,进行reference panel构建;二是可以省去构建reference panel步骤,参照STITCH等软件方法,通过大样本量低深度数据直接进行填充。
下节预告
下节将讲述填充准确性及分析方法,欢迎大家订阅。
参考文献
[1]Yang W , Yang Y , Zhao C , et al. Animal-ImputeDB: a comprehensive database with multiple animal reference panels for genotype imputation[J]. Nucleic Acids Research, 2019(D1):D1.
[2]Gao Y , Yang Z , Yang W , et al. Plant-ImputeDB: an integrated multiple plant reference panel database for genotype imputation[J]. Nucleic Acids Research, 2020.
[3]Peterson B K , Weber J N , Kay E H , et al. Double Digest RADseq: An Inexpensive Method for De Novo SNP Discovery and Genotyping in Model and Non-Model Species[J]. Plos One, 2012, 7:e37135.
[4]Mott, Richard, Flint, et al. Rapid genotype imputation from sequence without reference panels[J]. Nature Genetics, 2016.
近期活动

《报名开启 | 2021“单细胞与空间多组学技术应用”主题学术研讨会》(点击标题来报名参加吧~)