






2025年11月6日,兰州大学焦作义团队Cancer Research在线发表了题为“USP20-Driven Cholesterol Metabolism Links Inflammatory Signaling to Malignancy and Stromal Co-evolution in Pancreatic Cancer”的论文。本研究发现PDAC中,肿瘤固有炎症级联调控甲羟戊酸通路,驱动恶性进展与基质共进化,揭示了STAT3-USP20-HMGCR轴作为PDAC恶性进展的核心协调机制,并发现通过抑制USP20位点可抑制致癌信号、干扰代谢重编程并逆转微环境重塑,为PDAC的治疗提供新的靶点。欧易生物提供了空间转录组的测序工作。

发表期刊:Cancer Research
影响因子:16.6
涉及的欧易生物服务产品:空间转录组测序
研究背景
胰腺导管腺癌(PDAC)具有高度纤维化、免疫抑制性强的肿瘤微环境(TME),并且具有恶性程度高的特点。持续炎症信号、基质重塑和代谢重编程等促癌进展是PDCA恶性程度高的主要原因,但肿瘤细胞炎症程序如何协同调控致癌转化、基质重塑与代谢重编程仍不明确。USP20为已知的稳定HMGCR的去泛素化酶,近几年在肿瘤中的作用才获得关注。因此PDAC中炎症程序如何调控肿瘤进展以及USP20在癌症进展过程中的主要作用机制需要我们进一步研究,为开发更有效的PDAC治疗策略提供理论依据。
研究内容
本研究通过人的PDAC,KC-PC,KC-Ctr小鼠胰腺组织单细胞测序,揭示TNFSF13B+ 肿瘤相关巨噬细胞激活了肿瘤上皮细胞的STAT3信号通路,导致USP20的转录上调,并在空转数据中进行验证。代谢组学,发现炎症驱动的肿瘤甲羟戊酸代谢增强YAP/TAZ信号通路,进一步激活CAFs。之后通过构建类器官模型,动物实验模型来揭示PDAC恶性进展的核心机制为STAT3- USP20-HMGCR轴,并提出靶向抑制USP20作为抑制致癌信号、干扰代谢重编程并逆转微环境重塑的治疗策略。
技术路线

研究结果
1. 肿瘤内在炎症信号驱动肿瘤恶性进展和CAFs激活
为研究PDAC中的致癌信号网络,作者整合了人的PDAC和正常胰腺组织的scRNA-seq,发现炎症信号通路在癌症组织的肿瘤上皮细胞中显著上调,并发现STAT3的高表达与PDAC的不良预后相关(图1A~D);为模拟炎症驱动的PDAC进展,作者构建了患有(KC-CP)或不患有(KC-Ctr)慢性胰腺炎的KC小鼠,并对其胰腺组织进行scRNA-seq,InferCNV分析和功能富集结果显示,Malignant cell 1恶性最高,同时伴有炎症信号通路的激活和纤维化相关通路的增强(图1E~G);临床PDAC队列显示,STAT3高表达患者的胶原含量显著高于STAT3低表达组(图1H);作者通过空间转录组测序,发现KC-CP小鼠中CAFs和恶性细胞之间的细胞相互作用更强,空间共定位分析表明,与CAFs邻近的细胞中,恶性细胞占比最高,并且KC-CP小鼠的CAFs中胶原相关基因的表达升高,且与ECM相关的通路显著上调(图1I~L)。综上所述,肿瘤内在炎症信号促进CAFs激活,同时驱动PDAC的恶性进展和基质重塑。

图1 肿瘤内在炎症信号驱动肿瘤恶性进展和CAFs激活
2. TNFSF13B+巨噬细胞增加增强肿瘤细胞中STAT3介导的炎症信号
作者进一步探究恶性细胞中炎症信号的潜在机制,scRNA-seq分析发现KC-CP组中TNFSF13B+巨噬细胞的比例显著上调,空间转录组学数据进一步证实了这一点,功能富集发现KC-CP小鼠中TNFSF13B+巨噬细胞中的TNF信号通路显著上调(图2A~C);在人PDAC队列中发现,TNFSF13B+巨噬细胞富集的患者表现出更高的胶原沉积,pSTAT3表达也显著升高,且存活水平更低(图2D~H);并通过类器官培养实验,发现TNFSF13B可以增强类器官增殖,CAFs的胶原表达水平增高(图2I~J)。

图2 TNFSF13B+巨噬细胞介导的炎症信号传导促进肿瘤增殖和CAFs活化
3. 炎症驱动的肿瘤甲羟戊酸代谢增强YAP/TAZ介导的CAFs激活
作者通过构建小鼠胰腺组织和人类PDAC组织的类器官研究发现,CP类器官表现出STAT3磷酸化水平增加、STAT3调控基因表达升高、增殖能力增强,并且对共培养的CAFs具有更强的激活作用,转录组学分析发现,CP类器官表现出显著的ECM样和炎症样改变,且Hippo通路的关键基因CTGF和PAI-1的表达也显著上调(图2I~M);scRNA-seq进一步显示,Hippo通路在Malignant cell 1中富集,该簇与高恶性程度和高ECM得分相关(图2N)。
代谢组学分析显示,CP类器官中甲羟戊酸代谢的关键分支的活性增加,甲羟戊酸通路的关键代谢物(胆固醇和GGPP)在CP类器官中显著升高(图3A~B)。作者提出并通过湿实验来验证科学假说,即失调的甲羟戊酸代谢通过GGPP和胆固醇合成激活YAP/TAZ,促进PAI-1和CTGF的表达和分泌,并促进CAF激活和基质沉积(图3C~H)。为进一步研究甲羟戊酸代谢对PDAC基质沉积和肿瘤进展的影响,作者用HMGCR抑制剂辛伐他汀处理KC-CP小鼠和人类CP类器官发现,在KC-CP小鼠中,辛伐他汀降低了YAP/TAZ表达,抑制了基质沉积,并减少了肿瘤面积和分级(图3I~K);并在更具侵袭性的自发性PDAC小鼠模型(KPC小鼠)进行验证,发现HMGCR表达升高的PDAC中,胶原水平显著升高(图3L~P)。这些结果突显了HMGCR介导的甲羟戊酸代谢在促进PDAC基质沉积和恶性进展中的作用。

图3 HMGCR驱动的甲羟戊酸代谢促进了基质沉积以及胰腺导管腺癌的恶性发展
4. STAT3/USP20轴通过去泛素化HMGCR促进甲羟戊酸代谢和YAP/TAZ信号
为了进一步探究炎症是否通过去泛素化HMGCR促进甲羟戊酸代谢和YAP/TAZ信号,作者进行了HMGCR的免疫沉淀质谱分析发现,USP20是对照和CP类器官之间的差异表达基因唯一的去泛素化酶,USP20水平在CP类器官和KC-CP小鼠的胰腺组织中显著升高(图4A~E),Co-免疫沉淀和MST实验表明,USP20与HMGCR直接相互作用(图4F~G),泛素化实验和环己酰亚胺追踪实验显示,USP20抑制K48连接的多泛素化介导的HMGCR降解(图4H~I);人类PDAC样本显示,USP20和HMGCR蛋白质水平之间存在正相关(图4J);HMGCR敲除减弱了USP20缺失对YAP/TAZ信号的影响(图4K),证实了HMGCR在该轴中的核心地位。通过转录因子数据库预测,发现STAT3是USP20的潜在转录因子,并通过湿实验进行验证(图4L~O);在USP20缺陷类器官中,STAT3敲低不会降低HMGCR水平或抑制YAP/TAZ通路(图4P),这表明STAT3对HMGCR和YAP/TAZ的影响依赖于USP20。

图4 STAT3/USP20轴通过去泛素化HMGCR来促进YAP/TAZ的激活
5. USP20促进PDAC进展并损害ICIs的疗效
为评估USP20在PDAC进展中的作用,我们分析了临床样本中USP20的表达及其与预后的关系。结果显示,与邻近正常组织相比,PDAC肿瘤组织中USP20水平显著更高,且USP20高表达与不良存活结果相关(图5A~C)。此外,USP20表达升高的PDAC组织中,胶原水平显著更高(图5D)。USP20缺失显著抑制了CP类器官的增殖,破坏了YAP/TAZ信号,下调了CTGF和PAI-1的表达和分泌,并抑制了CAF激活(图5E~G)。此外,动物实验进一步表明,Usp20敲低增强了PD-1和/或CTLA-4抗体在KPC来源的同种异体移植小鼠中的治疗效果,并减少了肿瘤组织中的基质沉积和CD8+T细胞浸润(图5H~K)。这些结果表明,USP20是介导PDAC恶性进展和损害ICIs疗效的关键靶点。

图5 USP20可促进PDAC进展并降低ICIs疗效
6. GSK2643943A靶向USP20减少基质沉积、抑制恶性进展并增强ICIs的疗效
作者使用虚拟分子对接和MST实验测试了GSK2643943A其对USP20的亲和力,证实了两者之间的强相互作用(图6A~B)。泛素化实验表明,GSK2643943A抑制USP20对HMGCR的去泛素化活性(图6C)。用GSK2643943A处理可降低HMGCR水平,抑制YAP/TAZ通路,下调PAI-1和CTGF的表达,从而减少CAF激活并抑制CP类器官生长(图6D~G)。在KC-CP小鼠中,GSK2643943A减少了基质沉积,降低了肿瘤面积和分级,并抑制了肿瘤进展(图6H~K)。在KPC模型中,GSK2643943A显著抑制了基质沉积、PD-L1表达,上调了CD8+T细胞浸润,并增强了抗PD-1和抗CTLA-4抗体的疗效,并且GSK2643943A与抗PD-1和抗CTLA-4抗体联合使用可导致肿瘤显著消退(图6L~N)。
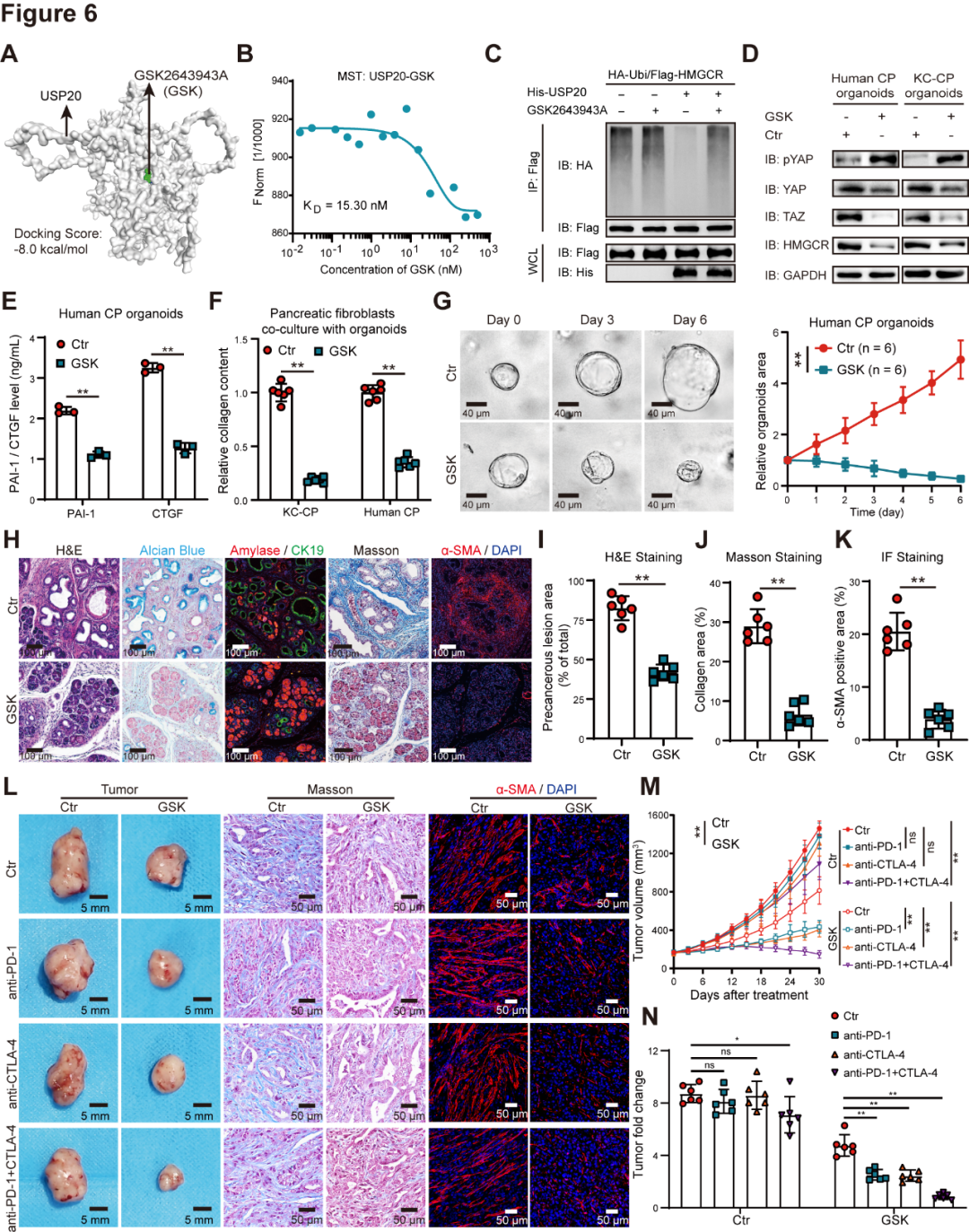
图6 GSK2643943A通过抑制基质沉积,可抑制PDAC进展并使ICIs疗效提升
7. GSK2643943A与抗PD-1/CTLA-4联合使用在PDAC临床前模型中表现出强效治疗效果
鉴于GSK2643943A与抗PD-1/CTLA-4联合使用具有强效治疗效果,作者使用KPC小鼠和来自3名患者的患者来源异种移植(PDX)模型,评估了这种联合治疗在PDAC中的长期疗效。在KPC模型中,GSK2643943A与抗PD-1和抗CTLA-4联合使用显著延长了KPC小鼠的存活时间(图7A~B)。为评估GSK2643943A与卡度尼利单抗联合使用的疗效,作者通过输注PBMCs重建了PDX小鼠的免疫系统,并进行了药物干预(图7C)。结果显示,在PDX-1、PDX-2和PDX-3中,GSK2643943A抑制了基质沉积和肿瘤生长(图7D-F),且其与卡度尼利单抗联合使用显著增强了治疗效果并延长了总生存期(图7G~J)。

图7 GSK2643943A联合抗PD-1/CTLA-4疗法可延长胰腺导管腺癌患者的总生存期
研究结论
1. PDAC中肿瘤内在的炎症级联反应是甲羟戊酸途径异常激活的主要调控因子,该途径既驱动恶性进展,又促进基质共进化。
2. STAT3-USP20-HMGCR轴是PDAC恶性进展的核心调控因子,并表明抑制USP20是一种可同时阻断致癌信号、干扰代谢重编程和逆转微环境重塑的治疗策略。
参考文献:
JJiang X, Zhao B, Wang T, Ma Y, Liu W, Sun H, Li Z, Wang K, He Q, Guan X, Qin L, Shi W, Dong Y, Ye Z, Zhou C, He X, Qing H, Long B, Zhou H, Yu Z, Jiao Z. USP20-Driven Cholesterol Metabolism Links Inflammatory Signaling to Malignancy and Stromal Co-evolution in Pancreatic Cancer. Cancer Res. 2025 Nov 6. doi: 10.1158/0008-5472.CAN-25-1228. Epub ahead of print. PMID: 41196022.