






胰腺导管腺癌(PDAC)恶性程度极高、5年生存率仅13%,免疫检查点抑制剂(如抗PD-1)疗效普遍不佳,核心瓶颈在于致密纤维化肿瘤微环境(TME):肿瘤相关成纤维细胞(CAFs)大量分泌细胞外基质(ECM),形成物理屏障并抑制CD8+T细胞浸润,导致免疫逃逸与治疗耐药。糖酵解异常是PDAC核心代谢特征,过量乳酸可诱导组蛋白乳酸化修饰,但乳酸化如何驱动基质沉积、调控免疫微环境的分子机制仍不明确,严重制约靶向策略开发。
2026年2月,兰州大学第二医院焦作义教授研究团队在Advanced Science(IF 14.1)发表题为UBE2T-Driven p53 Degradation Rewires Glycolysis to Orchestrate Lactylation-Mediated CAFs Activation and ECM Deposition in Pancreatic Cancer的重磅研究。该研究整合空间代谢组、Bulk RNA-seq、单细胞测序、CUT&Tag等技术,系统阐明UBE2T驱动p53正反馈降解、重编程糖酵解、调控CAFs乳酸化与ECM沉积的完整机制,并证实UBE2T抑制剂PGG联合抗PD-1可显著增敏免疫治疗,为PDAC提供全新靶点与联合方案。
【欧易生物为本研究提供空间代谢组学+单细胞转录组测序+CUT&Tag技术服务】

1. 乳酸介导CAFs的H3K18la修饰驱动PDAC基质沉积
研究团队首先利用自发性胰腺癌小鼠模型开展空间代谢组分析,结果发现胰腺组织中ECM富集区域的中心碳代谢和乳酸积累水平增强,体外实验进一步证实外源性乳酸能够有效促进肿瘤相关成纤维细胞分泌胶原蛋白,并上调CAF活化标志物与ECM合成相关基因的表达水平;乳酸处理还可显著提升CAF的整体乳酸化水平,其中组蛋白H3K18位乳酸化(H3K18la)变化最为显著,CUT&Tag分析显示H3K18la在乳酸处理后会大量富集于Acta2、Col1a1等基因的启动子区域,且相关富集基因主要参与细胞外基质调控过程,临床PDAC样本检测也验证了CAF中H3K18la的表达水平与肿瘤基质沉积程度呈显著正相关,而敲低乳酸化关键调控分子Ep300后,乳酸诱导的H3K18la升高与胶原蛋白分泌增加现象均被明显逆转,充分说明肿瘤微环境中乳酸累积可通过上调CAF的H3K18la水平,直接驱动胰腺癌CAF活化与细胞外基质沉积。
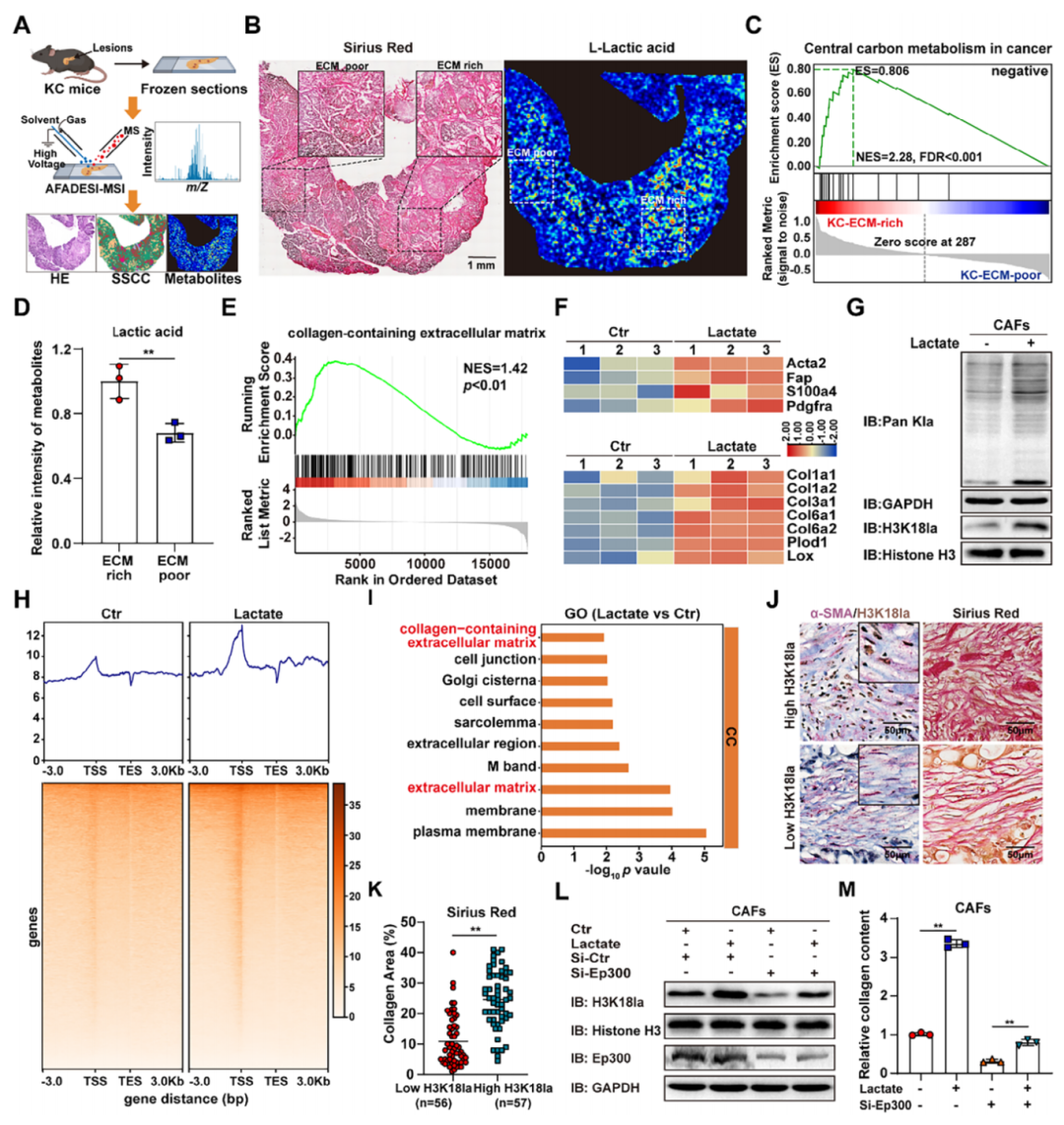
图1.CAF 中 H3K18 乳酸化驱动胰腺癌 CAF 活化与细胞外基质沉积
2. 抑制乳酸代谢可减轻基质沉积并恢复抗肿瘤免疫
为明确乳酸代谢对肿瘤微环境的调控作用,研究团队开展了靶向干预研究,结果发现抑制乳酸生成关键酶LDHA,能够有效降低CAFs中H3K18la水平并减少胶原分泌。通过单细胞转录组测序分析发现LDHA抑制后促纤维化、高免疫抑制活性的Dpp4+CAFs亚群比例明显下降,且该亚群的免疫抑制活性也显著降低;同时机体抗肿瘤免疫被全面激活,CD8+效应T细胞、NK细胞等免疫效应细胞比例提升,T细胞从幼稚态向效应态分化增强,CXCL16‑CXCR6信号轴介导的T细胞浸润效率提高。体内动物实验进一步证实,LDHA抑制剂联合抗PD-1治疗可有效降低肿瘤基质沉积、增强CD8+T细胞浸润,最终实现抑制肿瘤生长。综上证实,靶向乳酸代谢能够缓解胰腺癌基质纤维化并激活抗肿瘤免疫,提升免疫治疗效果。
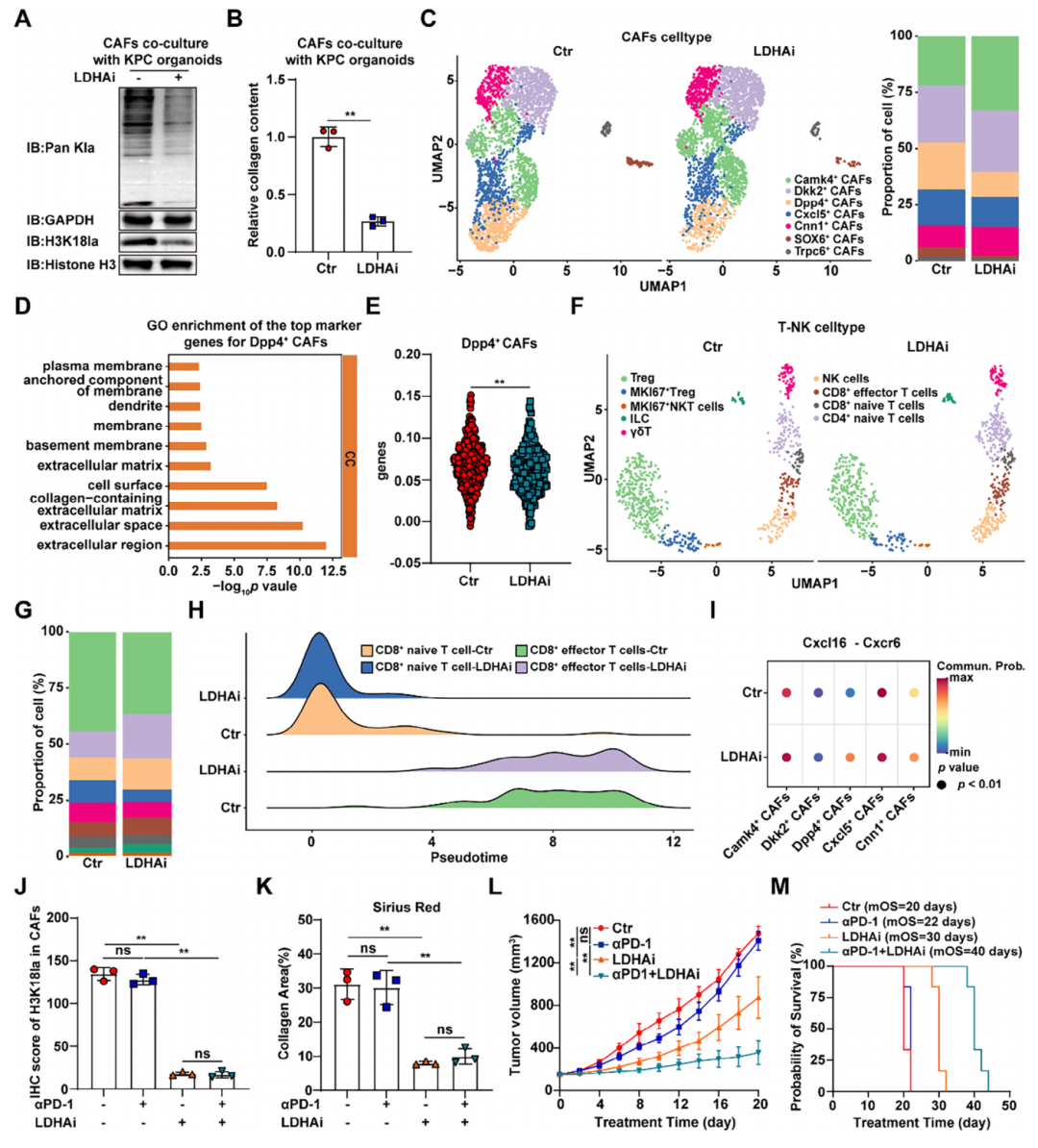
图2. 靶向乳酸代谢减轻基质沉积并激活抗肿瘤免疫
3. UBE2T驱动p53正反馈降解,促进CAFs乳酸化与基质沉积
为找到乳酸过量生成的上游核心开关,研究团队聚焦p53与泛素结合酶UBE2T展开深入探索,结果证实p53降解会直接导致肿瘤细胞乳酸分泌剧增,进而上调共培养CAFs的H3K18la水平并加速基质沉积。结合前期研究基础进一步探索上游调控机制,发现UBE2T正是启动这一过程的关键分子,它通过调控核糖体生物合成,增强核糖体蛋白L5(RPL5)与E3泛素连接酶MDM2的相互作用,最终促进MDM2介导的p53泛素化降解,并形成持续放大的正反馈循环,最终让肿瘤细胞糖酵解异常增强、乳酸大量释放。研究明确了UBE2T介导的p53正反馈降解是促进CAF乳酸化与基质沉积的核心分子机制。
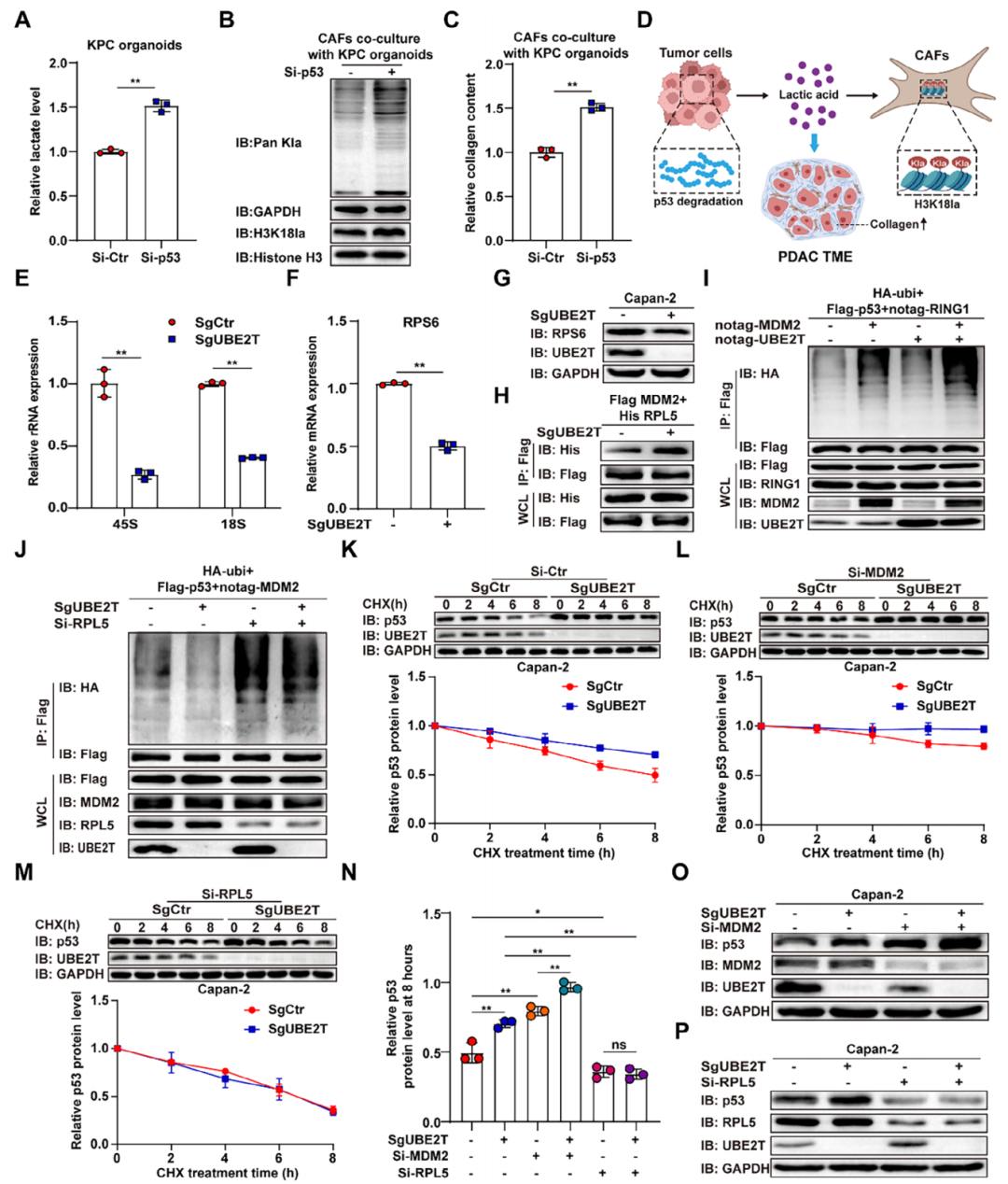
图3. UBE2T 驱动 p53 正反馈降解促进 CAF 乳酸化与基质沉积
4. UBE2T‑p53轴通过重编程糖酵解介导乳酸代谢串扰
为解析UBE2T对乳酸代谢的具体调控作用,研究团队进一步构建Ube2t条件敲除小鼠模型并开展了空间代谢组分析,结果显示敲除Ube2t后,鼠胰腺肿瘤区域与ECM区域的中心碳代谢活动减弱,乳酸水平显著下降。进一步实验发现UBE2T并非直接调控CAF的乳酸产生,而是通过肿瘤细胞与CAF之间的代谢对话发挥作用,肿瘤细胞中UBE2T缺失会降低自身糖酵解水平与乳酸分泌量,进而减少共培养CAF的乳酸累积、H3K18la水平及胶原蛋白分泌,而在肿瘤细胞中敲低p53可完全逆转UBE2T缺失带来的上述效应,证明UBE2T对乳酸代谢与基质沉积的调控依赖于p53通路。同时临床PDAC样本分析也证实,肿瘤细胞中UBE2T高表达与CAF中H3K18la高表达、肿瘤基质致密化呈显著正相关,充分阐明UBE2T-p53轴可通过重编程肿瘤细胞糖酵解过程,介导肿瘤细胞与CAF之间的乳酸代谢对话,最终驱动胰腺癌基质沉积。
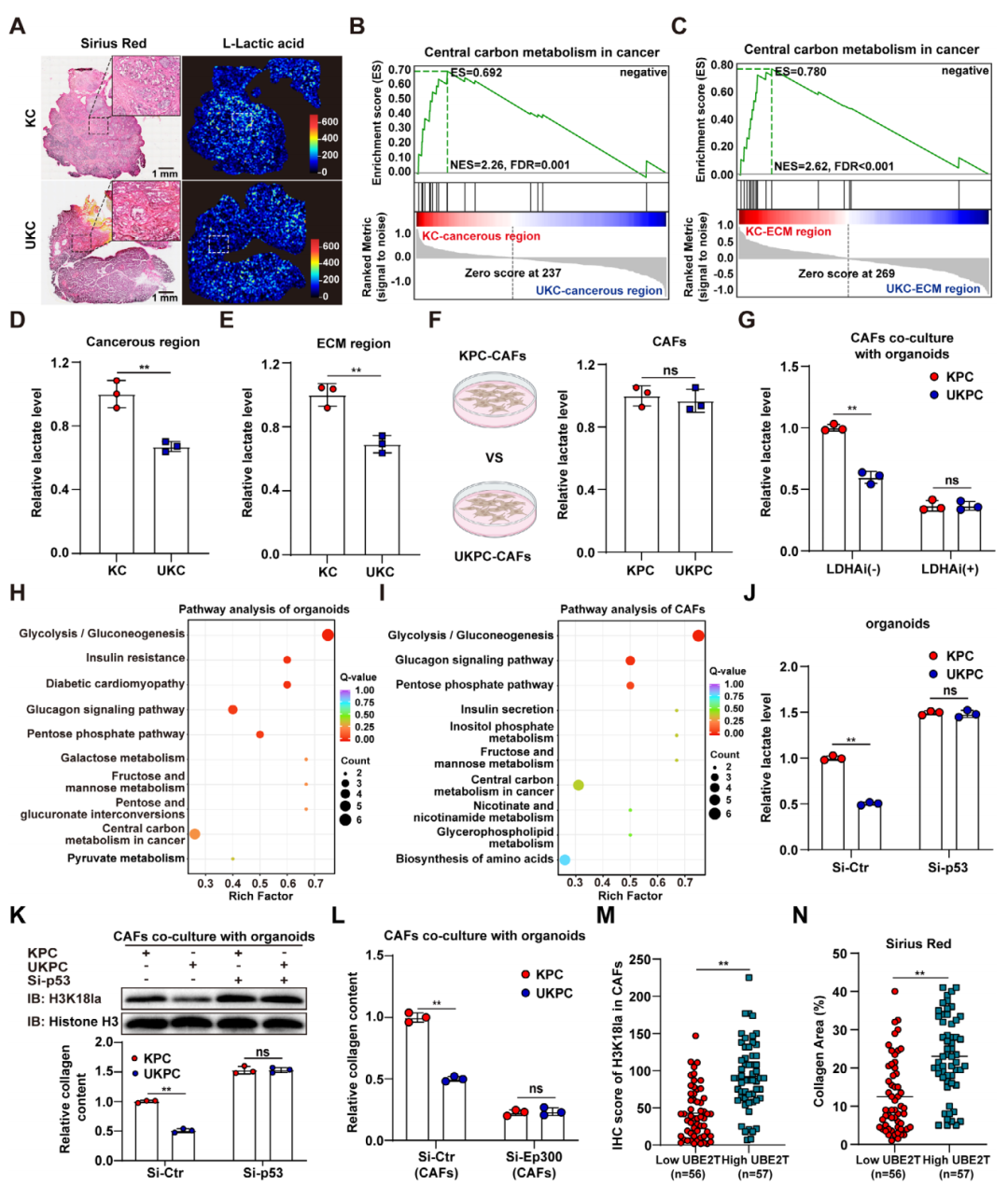
图4. UBE2T-p53 轴通过重编程糖酵解驱动乳酸代谢对话与基质沉积
5. UBE2T介导的基质纤维化严重损害抗PD-1免疫治疗疗效
为了评估UBE2T在肿瘤微环境中的调控作用及其对免疫治疗效果的影响,研究团队构建了KPC及Ube2t敲除的UKPC移植瘤模型,结果显示Ube2t敲除能够有效降低CAFs中H3K18la水平、减少ECM沉积,同时显著提升瘤内CD8+T细胞浸润数量。在KPC异种移植模型与Panc02细胞移植模型中均验证,Ube2t敲除联合抗PD-1治疗可显著抑制肿瘤生长、延长模型小鼠生存期,明确UBE2T是驱动胰腺癌基质致密化、导致免疫检查点抑制剂治疗抵抗的关键分子,靶向UBE2T可有效改善胰腺癌免疫治疗耐受状态。
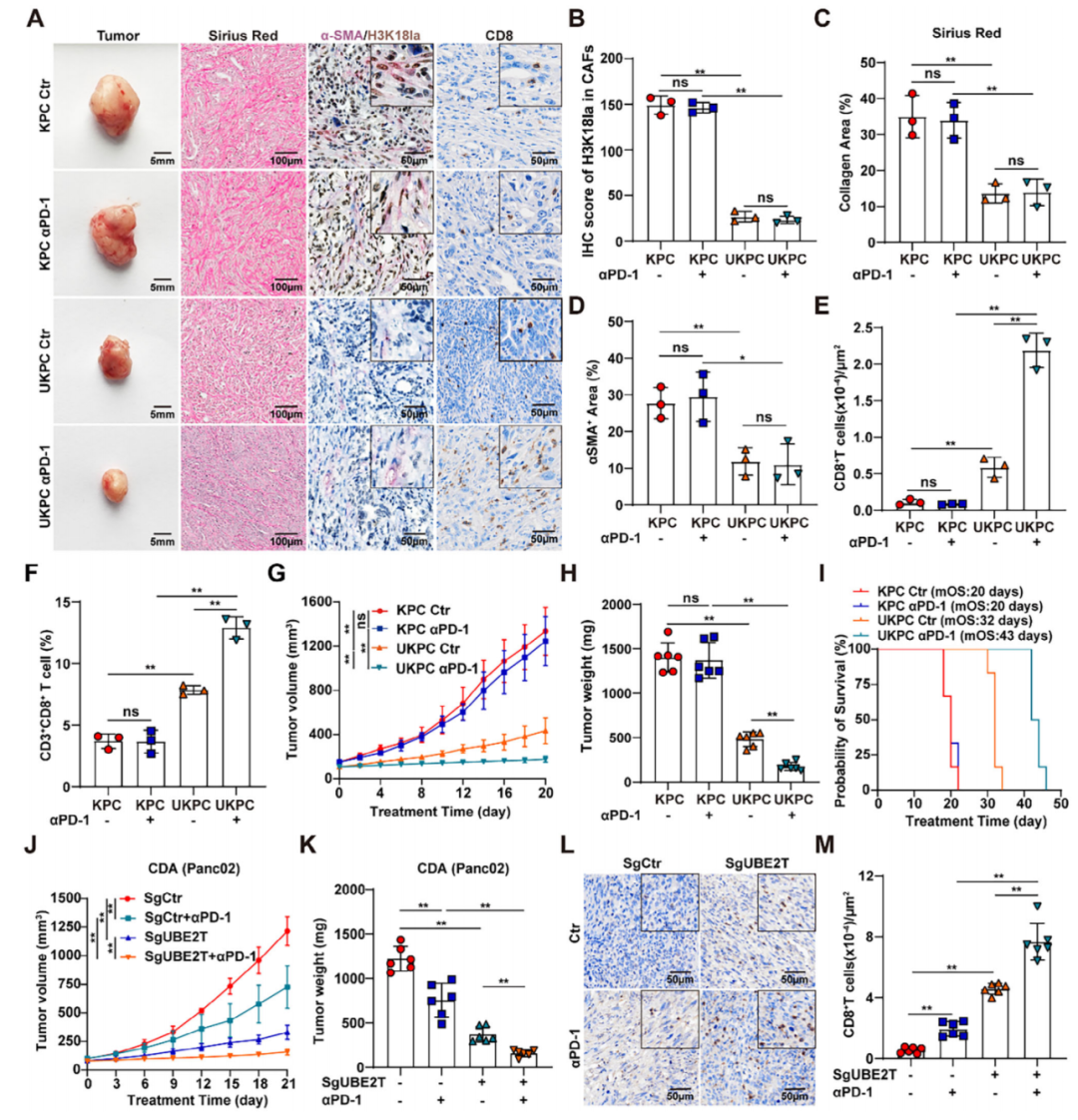
图5. UBE2T 驱动的基质增生损害胰腺癌抗 PD-1 免疫治疗疗效
6. PGG靶向抑制UBE2T,阻断乳酸代谢串扰与基质沉积
基于前期筛选的UBE2T特异性抑制剂五没食子酰葡萄糖(PGG),研究团队聚焦临床转化,对PGG开展系统功能验证。结果显示PGG处理可阻断异常核糖体生成,增强RPL5与MDM2的结合,进而抑制UBE2T驱动的p53正反馈降解。
将PGG处理后的KPC类器官与CAF共培养,发现PGG可同时降低肿瘤类器官与共培养CAF的糖酵解中间产物含量与乳酸水平,还能显著下调CAF的H3K18la水平并减少胶原蛋白分泌,而在Ube2t敲除的UKPC类器官共培养体系中,PGG未产生额外调控效应,证明PGG对乳酸代谢与基质沉积的抑制作用完全依赖于UBE2T靶点,证实PGG可通过靶向UBE2T有效阻断胰腺癌肿瘤细胞与CAF之间的乳酸代谢对话,抑制基质沉积。
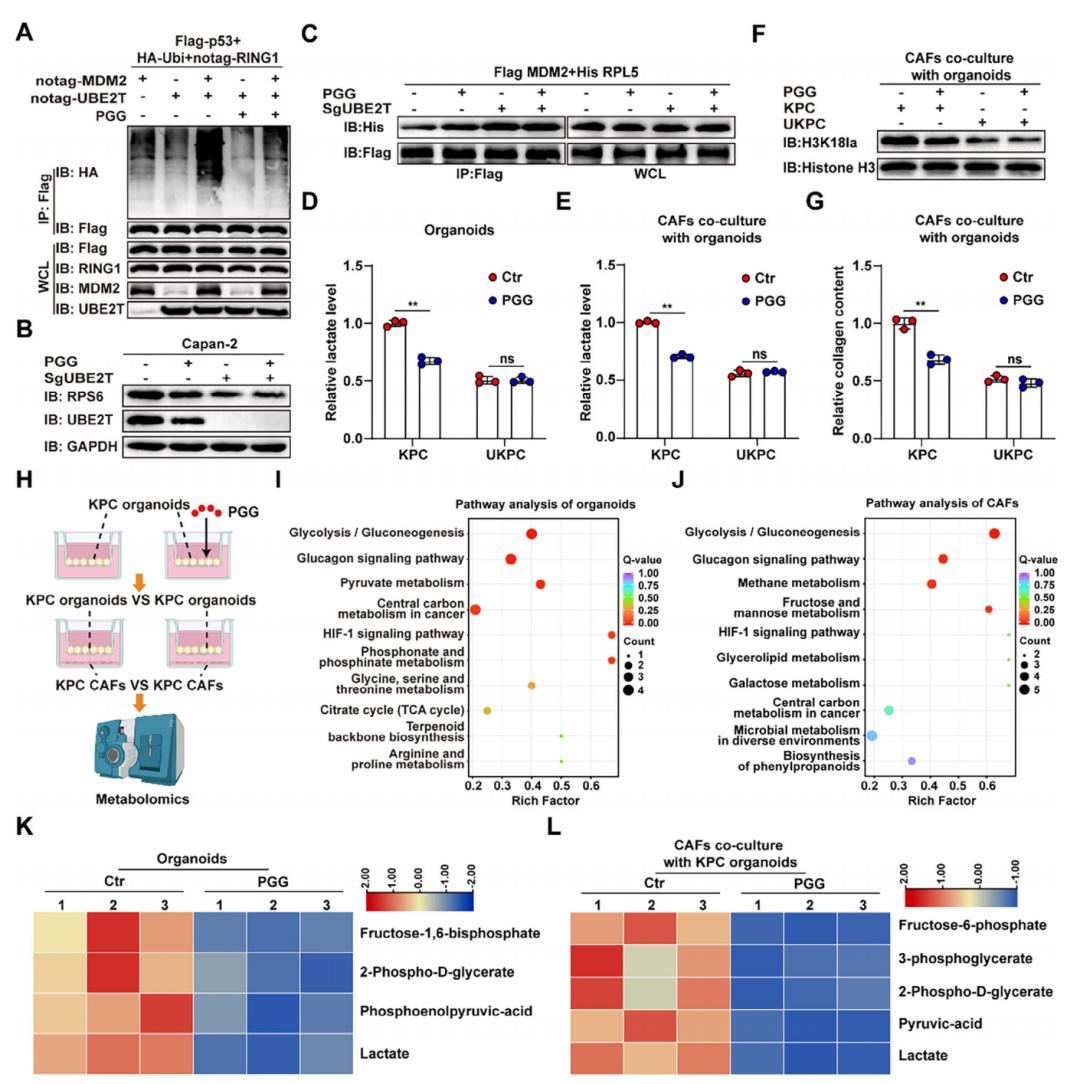
图6. PGG药理靶向UBE2T抑制乳酸代谢对话与基质沉积
7. PGG联合抗PD-1在PDAC模型中展现强效协同治疗效果
为了研究PGG对PDAC肿瘤微环境和免疫治疗效果的影响,研究团队用PGG和抗PD-1抗体处理KPC异种移植小鼠。结果显示PGG单药处理即可降低CAF的H3K18la水平、减轻肿瘤基质沉积、促进CD8+T细胞浸润,与抗PD-1抗体联合使用后呈现显著协同抗肿瘤效应。在更贴近临床的免疫重建人源PDX模型中,联合治疗同样表现出优异的抗肿瘤效果,还能大幅延长模型小鼠的中位生存期,充分证明PGG可作为胰腺癌免疫治疗增敏剂,与抗PD-1抑制剂联用成为极具前景的胰腺癌治疗新策略。
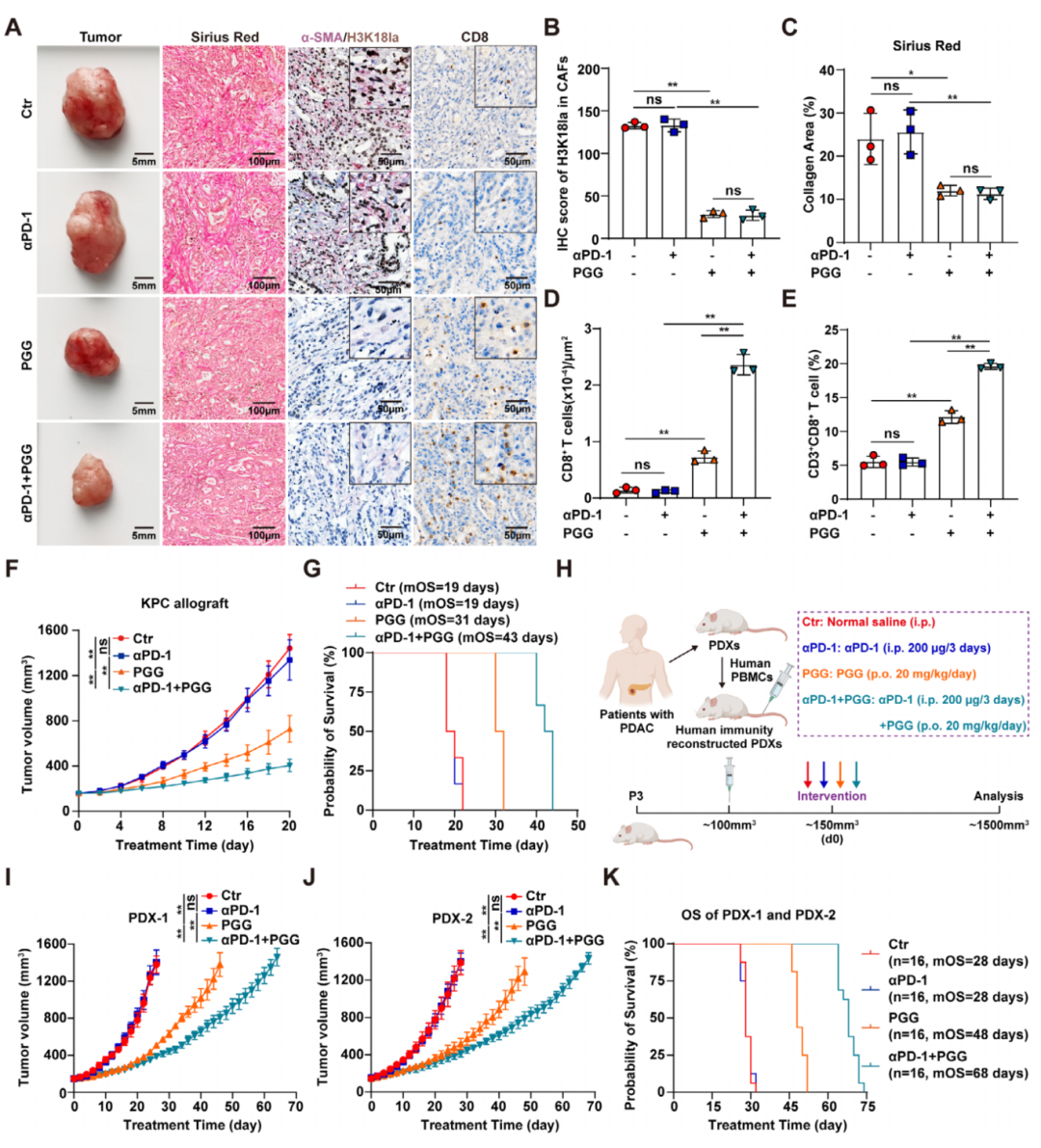
图7. PGG联合抗PD-1在胰腺癌中发挥强效治疗效应
本研究以UBE2T-p53-糖酵解-乳酸化-基质纤维化-免疫抑制为主线,完成从机制解析到靶点验证、再到联合治疗策略的全链条突破:阐明UBE2T是PDAC代谢重编程与微环境重塑的核心驱动因子,通过p53正反馈降解放大糖酵解与乳酸分泌;揭示乳酸-H3K18la是连接肿瘤代谢与CAFs活化、ECM沉积的关键表观调控环节;证实PGG靶向UBE2T可同时瓦解基质屏障、逆转免疫抑制,与抗PD-1联用实现强效协同抗肿瘤。该成果为攻克胰腺癌免疫治疗耐药提供了全新机制、关键靶点与可转化的联合治疗方案,具有重要科学价值与临床应用前景。