






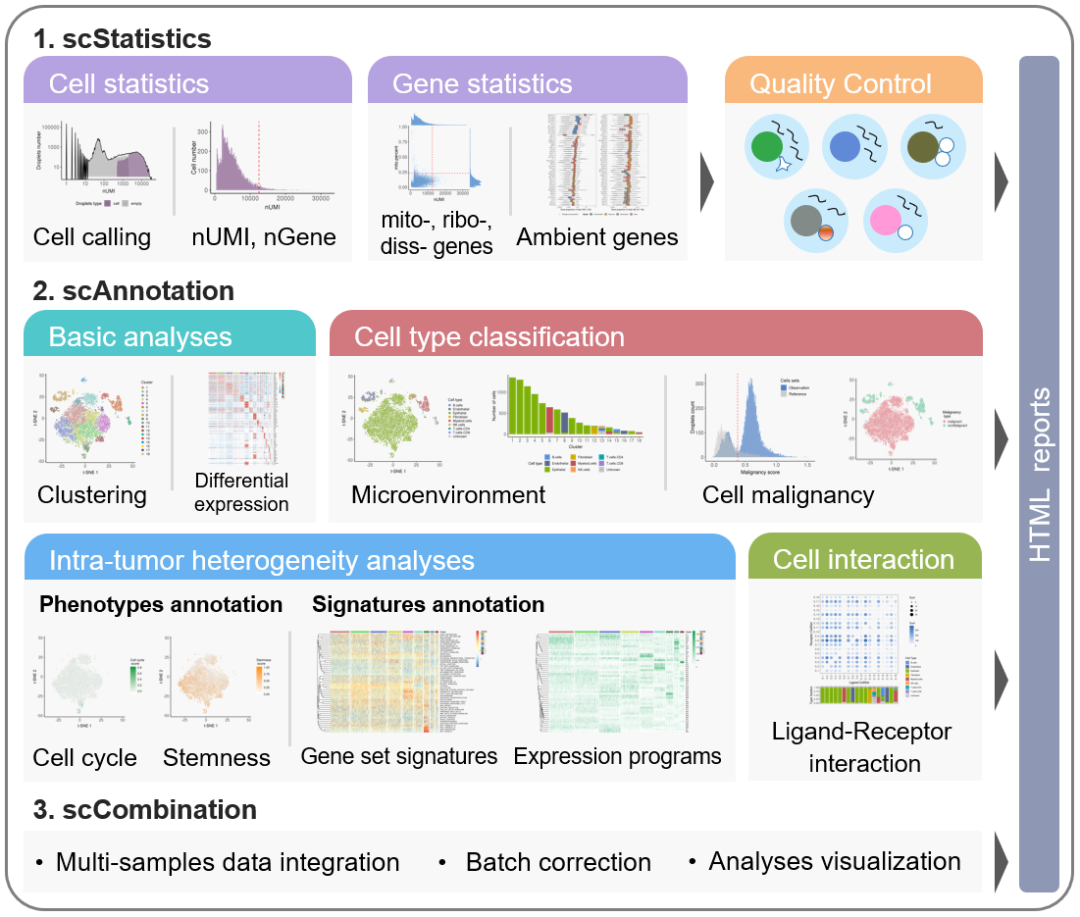
scCancer简介
癌症是全世界最致命的疾病之一,细胞异质性和复杂的微环境是当前癌症研究的主要挑战,而单细胞转录组测序 (scRNA-seq) 技术使得在单细胞水平上研究癌细胞的异质性及其微环境成为可能。
随着技术的发展,已有大量针对单细胞转录组数据分析的软件包被开发出来,极大促进了生物和医学领域的研究。然而,这些工具是为一般应用而设计的,缺乏对癌症特征的考虑,因此亟需要开发一款癌症特异性的分析工具,scCancer应运而生。
scCancer是2020年清华大学Jin Gu团队开发的一个用户友好型自动化 R 包,它专注于处理和分析用于癌症研究的 scRNA-seq 数据。该软件包括三个部分:一是数据统计(scStatistics),二是细胞注释(scAnnotation),例如降维聚类、细胞类型注释、恶性细胞估计、细胞表型分析、基因特征分析、细胞间相互作用分析等,三是多样本数据整合、批次效应校正和分析可视化(scCombination)。
该软件包可在以下网址获得:http://lifeome.net/software/sccancer/
本篇文章中,我们主要来学习在肿瘤研究中最为关注的恶性细胞评估和细胞干性分析。
scCancer原理
1.细胞恶性评估
在癌症研究中,有一个关键问题是估算细胞的恶性程度并区分出恶性和非恶性细胞。一般来说,从scRNA-seq数据中推断出的拷贝数改变 (CNV),具有识别恶性肿瘤细胞的潜力,可以广泛用于各种癌症类型的研究。在这里,作者使用了 R 包 infercnv 算法来估计单细胞 CNV。计算过程分为以下三步:
① 计算每个染色体上的移动平均表达值,然后将它们与正常的参考细胞进行比较,从而估算CNV。(软件包提供了正常细胞的参考数据集,也可以自行设置数据中的正常细胞作为参考。推荐后者,计算结果更为准确。)
② 此外,考虑到 dropout 的影响,作者建议使用细胞邻域信息来平滑 CNV 值,从而更好地区分恶性肿瘤。详细来说,利用聚类的邻域矩阵来识别每个单元 i 的邻居 j ∈ Nb(i),并使用归一化的相似度作为权重 w(∑w = 1)。通过计算每个单元 i 及其邻居初始 CNV 的加权平均值值 v(init)和v(init),得到最终的 CNV 配置文件 v(final)。
v(final) = 1/2 v(init) + 1/2 ∑wv(init)
③ 将染色体上最终 CNV 值平方的平均值定义为恶性肿瘤的评分。通过将恶性肿瘤评分的分布与参考细胞进行比较并检测其双峰性,为细胞分配恶性肿瘤标签。
2. 细胞干性分析
肿瘤内异质性在癌症样本中普遍存在,并且与癌症预后不良呈高度相关。作者使用 OCLR 模型训练了一个基于人的干/祖细胞数据集的干性特征基因集,此干性特征基因集和细胞表达向量之间的 Spearman 相关性系数,定义为干性得分。
目前软件支持人和小鼠,小鼠的基因集根据人的基因集同源转换得到。
了解完分析原理,接下来让我们一起看看分析结果吧~
结果说明
1. 细胞恶性评估结果
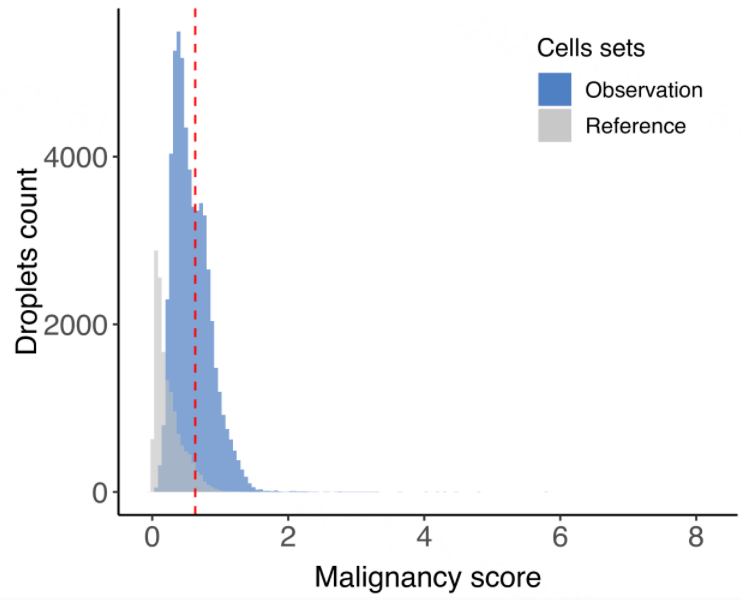
恶性评分分布直方图
说明:灰色为参考组的恶性评分分布情况,蓝色为观测组的恶性评分分布情况。红色虚线为识别的阈值,以区分恶性和非恶性细胞。高于此阈值的细胞判定为恶性细胞。
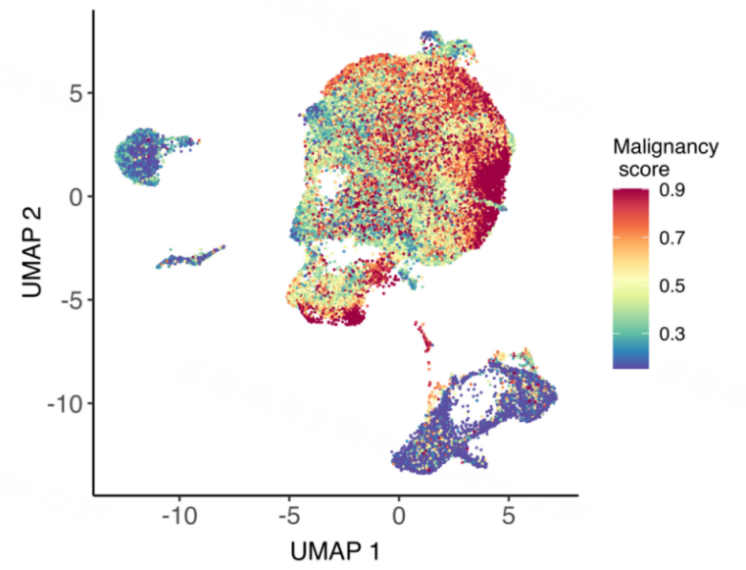
细胞恶性评分UMAP图
说明:UMAP图显示细胞的恶性肿瘤评分。颜色越红表明细胞的恶性程度越高。
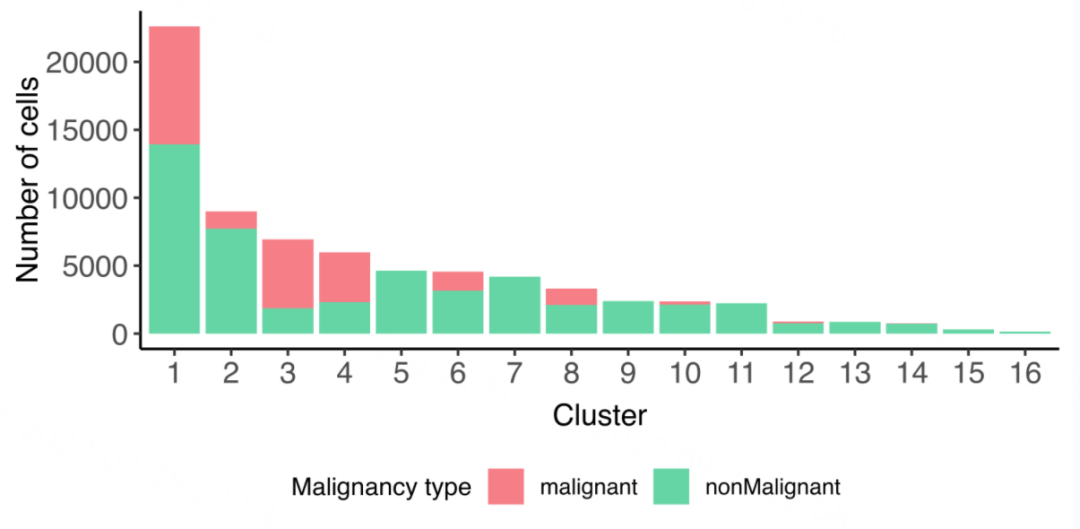
恶性和非恶性细胞统计柱状图
说明:红色为恶性细胞,绿色为非恶性细胞,横坐标为cluster,纵坐标为细胞数目。
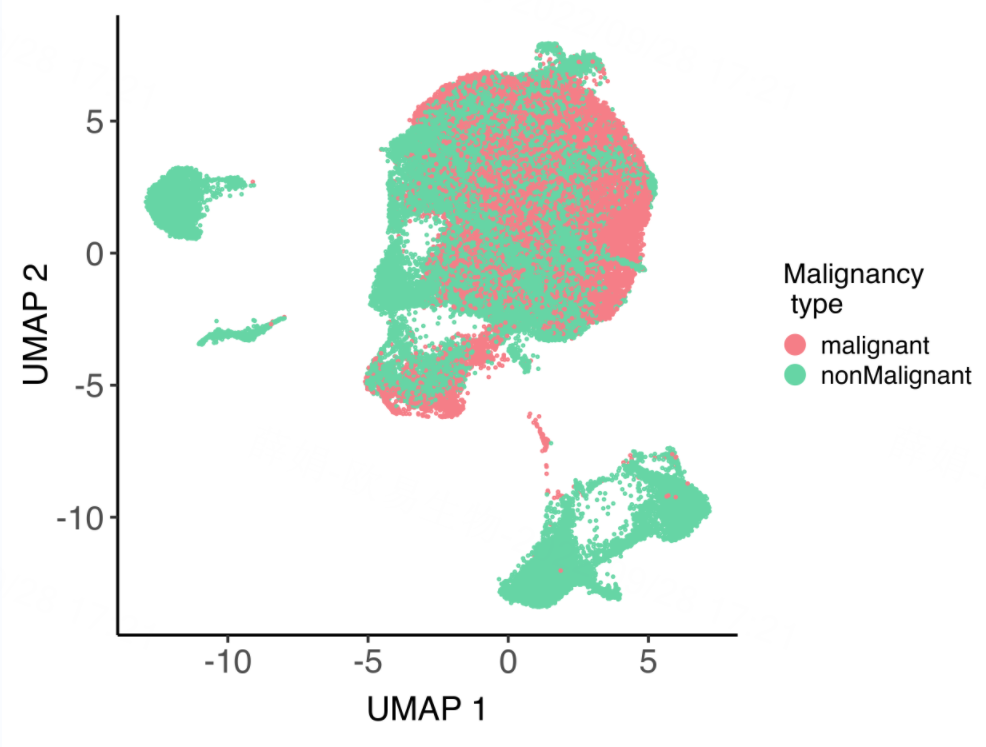
细胞恶性类型UMAP图
说明:红色表示恶性细胞,绿色表示非恶性细胞。
2. 细胞干性分析结果
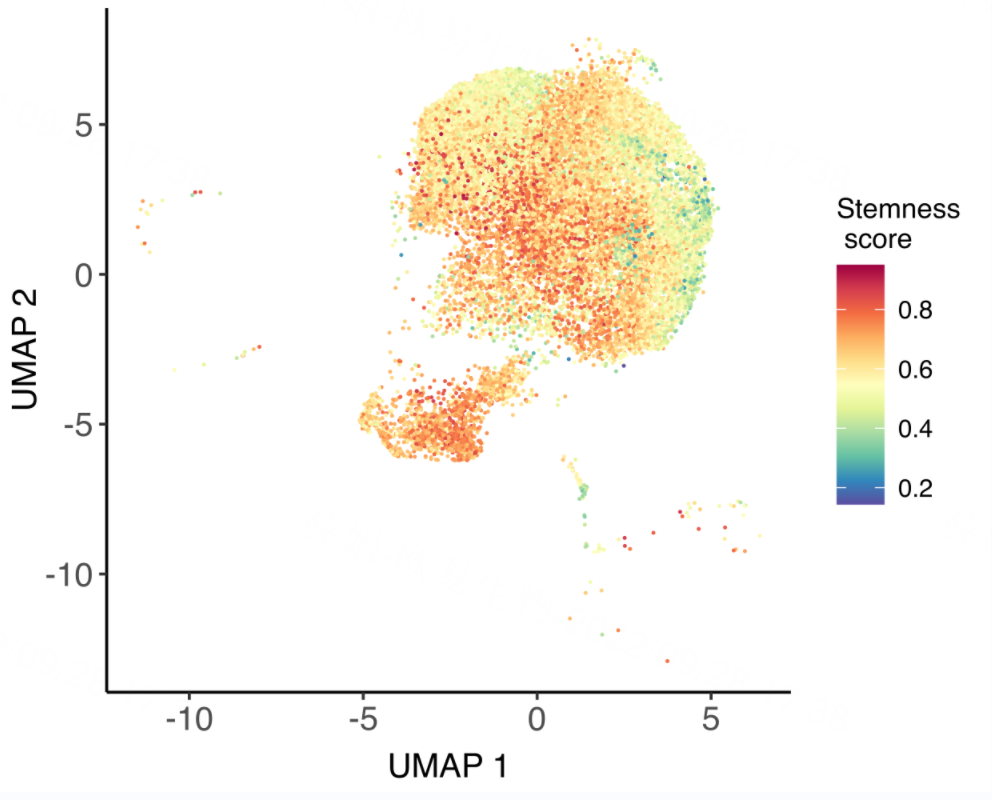
恶性细胞的干性评分UMAP图
说明:UMAP图显示恶性细胞的细胞干性评分。颜色越红表明细胞的干性程度越高。
参考资料:
1.Wenbo Guo, Dongfang Wang, Shicheng Wang, Yiran Shan, Changyi Liu, Jin Gu, scCancer: a package for automated processing of single-cell RNA-seq data in cancer, Briefings in Bioinformatics, bbaa127, https://doi.org/10.1093/bib/bbaa127
详细技术请访问欧易生物官网
⬇
百度搜索欧易生物(oebiotech)
⬇
了解更多多组学技术