






2025年5月14日,中南大学湘雅医院内分泌科,内分泌研究中心周海燕团队在著名期刊《Cellular & Molecular Immunology》在线发表了题为“Sexual dimorphism of lung immune-regulatory units imprint biased pulmonary fibrosis”的研究论文,该研究揭示了肺组织中免疫细胞基因存在显著的性别二态性,其中GCA表现出明显的性别差异,并通过其受体PTPRT促进致病性Th17细胞的募集,从而通过靶向GCA来预防和治疗性别特异性的肺纤维化。欧易生物参与了单细胞转录组测序相关工作。

发表期刊:Cellular & Molecular Immunology
影响因子:19.8
肺纤维化(PF)是一种慢性、进行性且不可逆的肺部疾病,男性患者的发病率、疾病进展速度以及死亡率均显著高于女性,但其机制尚不明确。本研究聚焦于肺组织中免疫细胞基因存在显著的性别二态性,阐明了PF中性别二态性的机制,其中颗粒钙蛋白(GCA)表现出明显的性别差异,证明了靶向GCA的中和抗体(GCA-NAb)显著减轻了雄性小鼠严重的PF病理,表明性别偏向的肺免疫调节单元能调控肺纤维化,并作为限制肺纤维化的靶点。

1.GCA在PF进展过程中表现出明显的性别二态性
为了证实肺纤维化存在性别差异,通过给8周龄小鼠气管内注射博来霉素(BLM)来构建肺纤维化动物模型,与雌性小鼠相比,在注射BLM后,雄性小鼠的肺功能显著受损,胶原沉积增加,肺呼吸功能下降(图1A-D)。在老年小鼠中,雄性小鼠比雌性小鼠更易患上严重的肺纤维化(图1E)。为了进一步探究性别差异背后的机制,我们对雄性小鼠进行了去势手术,发现雄激素剥夺对肺部病理几乎没有影响,而雌性小鼠卵巢切除的肺纤维化比假手术组小鼠更明显,但其严重程度仍低于雄性小鼠(图1F,G)。这些发现表明,性激素可能并非是导致PF严重程度主要决定因素。我们比较了雄性和雌性PF小鼠在注射BLM第7天后的支气管肺泡灌洗液 (BALF) 和骨髓 (BM) 免疫细胞的转录谱,从雄性BALF和BM中分离的免疫细胞转录谱存在显著差异,而雌性BALF与BM免疫细胞有相似的转录特征(图1H-J)。为了明确导致男性偏向性PF进展的途径和因素,我们进一步鉴定了在雄性PF小鼠的BALF和男性PF患者的肺免疫细胞中均高表达的基因,在27个共有基因中,基因Gca引起了我们的注意(图1K)。在未注射BLM时,雄性和雌性小鼠 BALF 中的GCA水平相似,但在注射BLM后,GCA在雄性小鼠的表达水平更高,且呈时间依赖性(图1L)。免疫荧光(IF)染色显示,GCA主要在纤维化肺组织内的巨噬细胞中表达(图 1M)。通过公共肺单细胞测序数据库分析可知,在年轻或老年男性的肺巨噬细胞中,GCA的表达也高于同龄的女性,且在PF患者中更为明显(图 1N)。总之,这些结果表明,肺存在性别特异性的GCA表达,这可能导致肺纤维化的性别差异。

图1 GCA表明肺免疫细胞在肺纤维化进展过程中存在明显的性别二态性
2.髓源性GCA加重肺纤维化并损害肺功能
为了阐明GCA对体内肺纤维化的影响,从BLM诱导肺纤维化的第一天起,雌性小鼠每周两次静脉注射重组GCA(rGCA)蛋白(图2A),GCA给药后小鼠存活率显著降低(图2B)。外源性GCA导致肺功能恶化,表现为气道阻力增加、每分钟通气量和依从性降低(图2C-E)。通过计算机断层扫描(CT)成像评估肺形态,表明 rGCA治疗组肺部病理恶化更严重(图2F)。通过HE、Masson和α-SMA染色表明,GCA治疗增加了胶原沉积(图2G)。这些发现共同表明GCA会加剧小鼠的肺纤维化。为了进一步明确巨噬细胞来源的GCA在肺纤维化发生中的作用,将Gca-floxed小鼠(Gcaf/f)与Lyz2-Cre小鼠杂交,构建髓源特异性GCA缺失的小鼠(GcaLyz2-KO)。我们分析了BLM诱导后PF相关表型(图2H),结果表明,雄性和雌性GcaLyz2-KO小鼠的PF显著改善,表现为生存周期延长(图2I)和呼吸功能的提升(图2J-L)。CT成像也显示两性小鼠肺功能均得到改善(图2M),并且GcaLyz2-KO小鼠的胶原沉积显著减少(图2N)。这些结果表明,髓源性GCA在肺纤维化的发展中起着至关重要的作用。

图2 髓源性GCA加重肺纤维化并损害肺功能
3.性别偏向的HTR2C+AMs调节GCA的产生并参与肺纤维化
我们接下来探讨了GCA的表达是如何以性别依赖的方式被调控的,在第7天,从PF两性小鼠中分离肺组织进行单细胞转录组测序。结果表明,与雄性小鼠相比,雌性小鼠肺组织中巨噬细胞的比例显著增加,而中性粒细胞的比例显著减少(图3A)。进一步分析巨噬巨噬细胞亚群,发现簇5在雌性小鼠中显著增加(图3B)。通过特征基因筛选发现Htr2c在簇5巨噬细胞中高度特异性表达(图3C),该亚群表现为CD11blowsiglecFhi表型,符合肺泡巨噬细胞(AMs)的特征(图3D),因此定义为HTR2C+AMs。GSVA通路富集分析显示,HTR2C+AMs中PPAR信号、脂肪酸降解和脂肪分解通路显著激活(图3E),这与AMs偏好利用脂质代谢供能的特性一致。此外,流式细胞术和IF染色表明,HTR2C+AMs在雌性小鼠的BALF和肺组织中高度富集,而在雄性中几乎不存在(图3F-H)。 为探究HTR2C+AMs是否调控GCA表达并影响PF进展,我们从雌性小鼠中分选出HTR2C+AMs和HTR2C-AMs,并通过气管内转移至野生型(WT)雄性受体小鼠体内(图3I)。与接受HTR2C-AMs的小鼠相比,HTR2C+AMs转移的小鼠BALF和肺组织中的GCA表达水平显著降低(图3J,K),并且接受HTR2C+AMs小鼠的胶原沉积减少和α-SMA表达降低(图3L)。这些结果表明,雌性偏向的HTR2C+AMs限制了GCA的产生并抑制了PF的进展。体外实验进一步验证,无论有无LPS刺激,与HTR2C+AMs共培养或使用其条件培养基(CM)处理,均可抑制巨噬细胞中GCA的表达(图3M-Q)。因此,AMs中的HTR2C对于抑制GCA的产生和肺纤维化的进展至关重要。
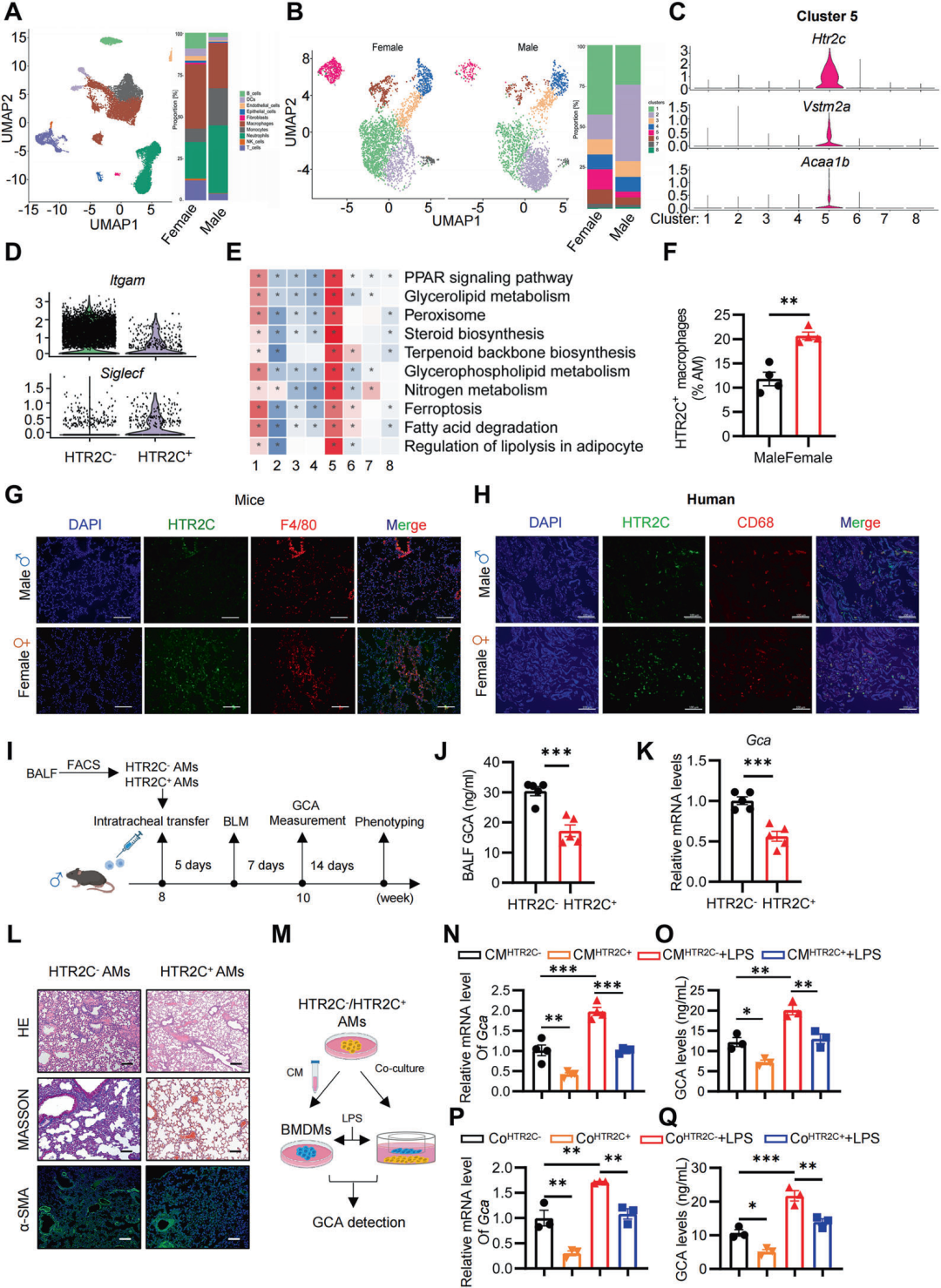
图3 性别偏向的HTR2C+AMs调节GCA的产生并参与肺纤维化
4.HTR2C+AM通过诱导代谢重编程来抑制巨噬细胞中的GCA产生
为了探究HTR2C+AMs影响浸润巨噬细胞中GCA表达的机制,通过转录组测序分析HTR2C- 或HTR2C+ CM培养的巨噬细胞,GSEA富集分析发现HTR2C+AMs 的CM激活了巨噬细胞的脂肪分解途径(图4A),这与HTR2C+AMs中甘油脂代谢和脂肪分解调控通路的富集结果一致(图3E)。无论是否经LPS处理,HTR2C+AMs的CM均能提高氧化磷酸化(OXPHOS),表现为基础耗氧率(OCR)和最大OCR的升高(图4B)。此外,HTR2C+ CM可逆转LPS诱导的代谢异常,恢复脂肪分解及脂肪酸氧化(FAO)相关基因的表达,HTR2C+ CM通过上调脂肪酶ATGL的mRNA和蛋白水平来抵消LPS对ATGL的抑制作用(图4C,D)。基于脂肪分解与糖酵解的拮抗关系,我们提出假说:HTR2C+AMs可能通过ATGL介导的脂肪分解抑制糖酵解,从而调控GCA+巨噬细胞的炎症扩增。siRNA敲低巨噬细胞中Atgl后(图4E),HTR2C+ CM诱导的代谢重编程被完全消除(图4F-H),且GCA表达的抑制效应显著减弱(图4I,J)。为了进一步证明代谢重编程在体内调控GCA扩增和PF进展中的作用,在Gca-Cre-GFP小鼠中,通过AAV-FLEX-shAtgl(Atgl GCA-KD)特异性敲低肺浸润GCA+巨噬细胞的Atgl基因(图4K)。BLM诱导7天后,Atgl GCA-KD小鼠中HTR2C+AM移植对BALF及肺组织GCA水平的抑制作用被显著抵消(图4L,M)。同时,Atgl缺失完全阻断了HTR2C+AMs对PF进展的保护(图4N)。综上所述,性别偏向的HTR2C+AM通过促进巨噬细胞代谢重编程(向脂肪分解和OXPHOS转化),抑制GCA的炎症扩增,从而延缓肺纤维化进展。

图4 HTR2C+AMs通过诱导代谢重编程抑制巨噬细胞中GCA的产生
5.GCA通过与其受体PTPRT结合诱导Th17细胞迁移
为进一步研究了GCA如何加速PF的进展,在BLM给药后第7天,RNA-Seq分析结果显示,GcaLyz2-KO组小鼠肺组织显著富集的通路与免疫功能相关,特别是Th17细胞分化和趋化因子信号通路(图5A)。尽管雄性小鼠的BALF中Th17细胞的比例高于雌性,但Gca缺失后两性的Th17细胞比例均显著降低(图5B)。qPCR分析进一步证实,Gca Lyz2-KO小鼠肺组织中Th17细胞特征相关基因的表达显著降低(图5C)。通过transwell迁移实验表明,GCA以剂量依赖的方式特异性诱导Th17细胞迁移,而其他辅助性T细胞亚群表现出更低的迁移趋势(图5D,E)。为了解析GCA诱导Th17细胞迁移的机制,通过质谱分析筛选Th17细胞中GCA的潜在受体,在6个候选受体中,蛋白酪氨酸磷酸酶受体T(PTPRT)在Th17细胞中表达显著高于其他T细胞亚群(图5F-I)。分子对接分析表明,GCA和PTPRT可以通过多组氢键结合形成稳定结构(图5J)。共免疫沉淀(co-IP)实验也证实了GCA与PTPRT相互作用(图5K)。为了确定负责相互作用的GCA关键结构域,通过系列GCA缺失突变体鉴定出51-86、92-127和158-193氨基酸区域为PTPRT结合必需域(图5L)。为了验证PTPRT介导GCA对Th17细胞迁移的机制,我们构建了CD4+T细胞特异性的Ptprt缺失小鼠(Ptprt CD4-KD)。IF染色显示,Ptprt WTTh17细胞结合His标记的rGCA,而Ptprt CD4-KDTh17细胞结合能力完全丧失(图5M)。Transwell实验表明,PTPRT缺失使Th17细胞对GCA的迁移能力受到抑制(图5N)。此外,μ-Slides趋化性实验表明,Ptprt WTTh17细胞呈现明显的GCA梯度定向迁移,而敲除组没有此现象(图5O)。
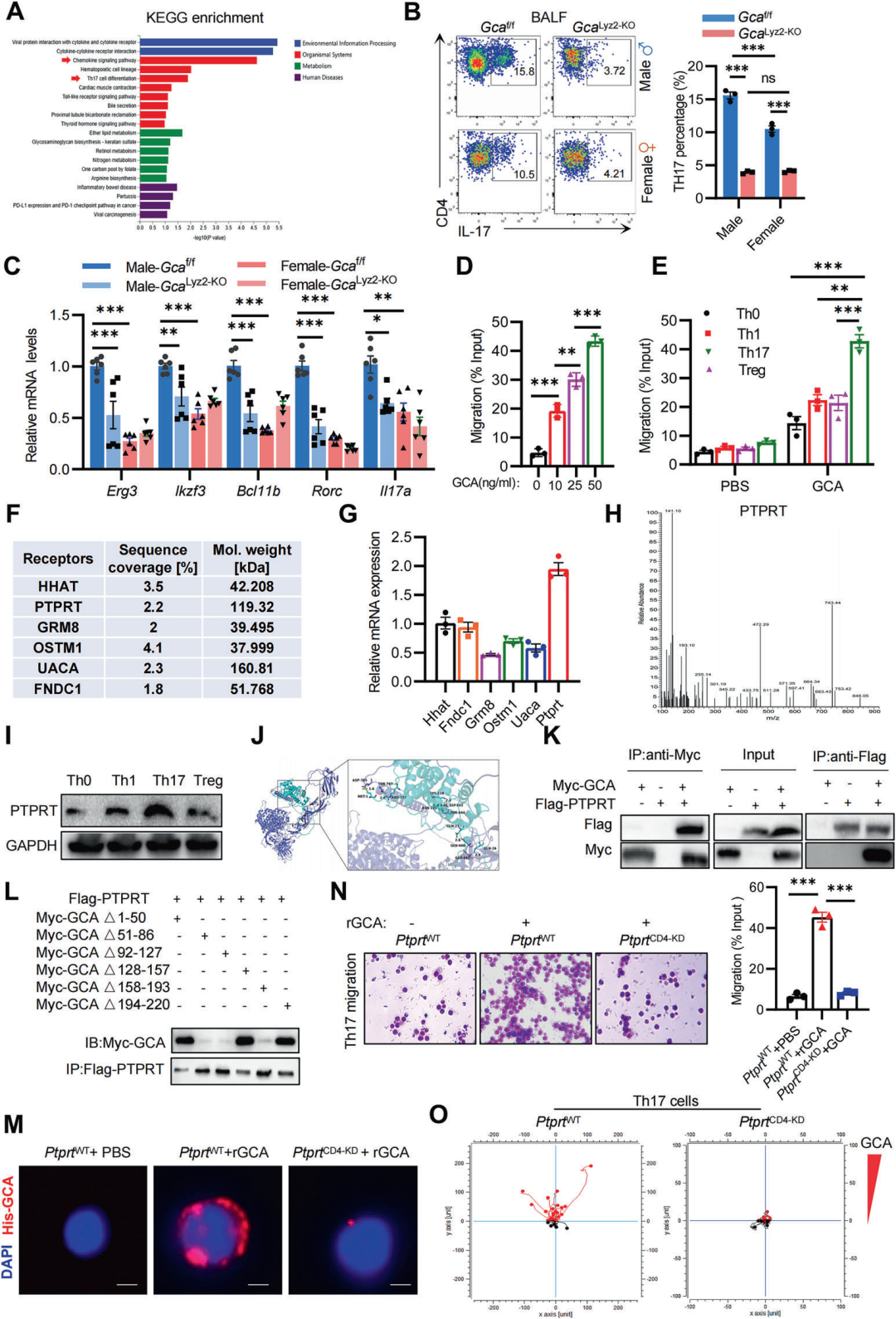
图5 GCA通过与其受体PTPRT结合诱导Th17细胞迁移
6.PTPRT消融可消除GCA诱导的Th17细胞迁移和肺纤维化
为明确PTPRT在GCA诱导的Th17细胞迁移及肺纤维化中的作用,在rGCA存在下进行BLM给药处理后第7天,我们对Ptprt WT和Ptprt CD4-KD雌性小鼠进行细胞迁移检测(图6A),与WT小鼠相比,Ptprt CD4-KD小鼠BALF和血液中Th17细胞的比例显著降低,而脾脏和纵隔淋巴结中的Th17细胞增加(图6B),结果表明Ptprt缺失阻断了GCA驱动的Th17细胞迁移。然而,Ptprt CD4-KD小鼠组织中的其他Th细胞亚群迁移无显著变化(图6C)。IF染色显示,过继性转移24小时后,受体小鼠肺中的Ptprt-/-Th17细胞数量显著降低(图6D),表明PTPRT直接调控Th17细胞的趋化性。rGCA刺激显著促进ROCK1-MLC通路磷酸化,而效应被PTPRT敲除消除且p-JNK不受影响(图6E),这表明GCA-PTPRT通过ROCK1-MLC通路调控Th17细胞的迁移。此外,ROCK1抑制剂Y27632可阻断rGCA诱导的Th17细胞迁移(图6F)。为了探究GCA如何刺激ROCK1激活,我们发现rGCA显著抑制Th17细胞中MYBPH(而非ARHGAP18/26)表达,该作用依赖PTPRT(图6G),并且MYBPH过表达可逆转rGCA诱导的ROCK1-MLC激活(图6H)。接下来研究PTPRT如何介导MYBPH的下调,发现GCA抑制了Th17细胞中的ERK1/2磷酸化(图6I),表皮生长因子(EGF)刺激ERK1/2可恢复MYBPH表达(图6J)。ERK1/2激活缓解GCA对转录因子TTF-1的抑制,siRNA敲低Ttf-1则抑制ERK1/2对MYBPH的诱导作用(图6K),表明GCA-PTPRT通过ERK1/2-TTF-1级联抑制MYBPH。最后,通过表型分析发现,Ptprt CD4-KD小鼠肺纤维化显著减轻,表现为胶原沉积减少及COL1A1和α-SMA表达降低(图6L,M)。综上所述,GCA通过结合PTPRT触发ERK1/2-TTF-1-MYBPH信号轴,激活ROCK1/MLC通路促进Th17细胞迁移,最终加速PF进展。

图6 PTPRT消融可消除GCA诱导的Th17细胞迁移和肺纤维化
7.GCA中和抗体显著缓解肺纤维化
我们证实GCA在小鼠肺纤维化进展中发挥核心调控作用。通过临床样本分析显示,相较于年龄匹配的女性患者,男性PF患者的病变组织中GCA+巨噬细胞数量显著增加,Th17细胞浸润水平升高(图7A-C),GCA+巨噬细胞比例与Th17细胞比例呈正相关(图7D),表明GCA-Th17轴参与疾病进展。在BLM诱导的肺纤维化模型中,每周两次给予GCA-NAb,21天后检测发现,显著改善了小鼠的呼吸功能(图7E-G),CT影像和HYP检测显示纤维化程度减轻(图7H,I),并且BALF中Th17细胞比例下降(图7J),这些结果表明GCA-NAb在预防和控制PF方面有着巨大潜力。GCA-NAb与吡非尼酮联用表现出更显著的疗效,表现为胶原沉积抑制效果增强(图7K-M),生存周期较单药治疗组显著提高(图7N)。综上所述,GCA-NAb单药治疗及与吡非尼酮联合方案均能有效缓解肺纤维化进展,具有重要临床转化价值。

图7 GCA中和抗体显著减轻肺纤维化
本研究首次揭示了肺免疫调节单元在塑造性别免疫二态性中的核心作用,阐明该单元是导致肺纤维化性别差异的核心机制,并证实GCA是一种新型Th17趋化因子,这些发现为开发靶向GCA的性别特异性肺纤维化防治策略提供了重要理论依据。
参考文献:
https://doi.org/10.1038/s41423-025-01293-8