







前言

2026年3月12日,安徽医科大学第一附属医院梅斌副教授/刘学胜教授团队,联合伦敦帝国理工学院/浙江大学马大青教授团队在Journal of Advanced Research(IF=13)上发表了题为“Locus coeruleus-hippocampus noradrenergic activation alleviates sepsis-associated encephalopathy by promoting astrocytic AQP4-related autophagy via α2A-AR”的文章,该研究深入解析了激活蓝斑-海马去甲肾上腺素能系统可以通过星形胶质细胞的α2A-AR,促进AQP4相关的自噬,从而减轻脓毒症相关脑病的核心机制,为开发针对脓毒症相关脑病的靶向疗法提供了新的理论依据和潜在策略。欧易生物提供了单细胞测序(scRNA-seq)的技术服务。


研究背景

脓毒症相关脑病(SAE)是脓毒症患者常见且严重的中枢神经系统并发症,发病率高,其导致的神经认知功能障碍显著增加患者的致残和死亡风险。尽管炎症诱导的中枢神经损伤被认为是SAE的关键机制,但具体机制尚未明确。蓝斑(LC)作为脑内去甲肾上腺素(NA)的主要来源,在调节神经认知功能中起重要作用。脓毒症可导致LC神经元凋亡,提示去甲肾上腺素能神经传递受损可能参与SAE的发生发展。α2A肾上腺素能受体(α2A-AR)是NA受体的重要亚型。作者前期研究发现,激活海马星形胶质细胞上的α2A-AR能够抑制星形胶质细胞过度反应性,从而改善脓毒症小鼠的长期神经认知功能,但其内在机制有待阐明。水通道蛋白-4(AQP4)在星形胶质细胞中高表达,不仅参与水稳态维持,也调控细胞反应性,并可影响自噬水平——后者是决定星形胶质细胞反应性的关键因素。值得注意的是,AQP4的表达受cAMP/PKA信号通路调控,而该通路同样是α2A-AR激活后的主要下游途径。深入阐明LC-HP-NA系统及星形胶质细胞α2A-AR在SAE中的作用与机制,有望为开发针对去甲肾上腺素能系统和星形胶质细胞反应的干预策略提供理论依据,从而为减轻脓毒症相关神经认知后遗症提供新的治疗靶点。

研究内容

本研究证实激活LC-HP-NA系统可减轻脓毒症诱导的长期神经认知损伤,并阐明了其潜在机制。综合运用sc-RNA-seq、化学遗传学及微透析、蛋白质印迹、免疫荧光等技术,系统阐明了LC-HP-NA系统激活对SAE的保护作用与机制。首先,通过sc-RNA-seq、行为学测试及微透析等,证实脓毒症损伤LC-HP-NA系统,导致海马去甲肾上腺素释放减少、星形胶质细胞AQP4表达上调、自噬功能受损及反应性增强,并引起长期认知功能障碍。进而,利用化学遗传学特异性激活LC-HP-NA,可逆转上述异常,改善认知功能,其效应依赖于星形胶质细胞α2A-AR。通过星形胶质细胞特异性敲低α2A-AR或过表达AQP4实验,证实二者是LC-HP-NA发挥神经保护作用的关键介质。最后,在LPS刺激的原代星形胶质细胞模型中,结合药理学干预与信号通路分析,阐明α2A-AR通过抑制cAMP/PKA信号下调AQP4,进而激活PPAR-γ/mTOR依赖性自噬、抑制细胞反应性的分子机制。

技术路线



研究结果

Result 1 脓毒症诱导海马星形胶质细胞、小胶质细胞和神经元的转录组变化
基于海马组织的单细胞转录组分析(Fig. 1a),作者研究发现脓毒症小鼠海马星形胶质细胞(Marker如Gfap、Slc1a3等,Fig. 1b)表现出显著的A1型星形胶质细胞活化特征,表现为Fkbp5、Fbln5等A1型活化相关基因表达上调(Fig. 1c)。同时,自噬相关基因Sqstm1(编码P62)和Map1lc3b(编码LC3B)的转录增加,提示自噬流可能受阻,而非激活。此外,水通道蛋白AQP4的表达也升高(Fig. 1c)。进一步聚类分析将星形胶质细胞分为五个亚群(Fig. 1d-f)。值得注意的是,脓毒症后所有亚群中A1活化基因的表达均普遍增强(Fig. 1g)。在占主体的簇1-4中,AQP4及自噬相关基因的表达也同步升高。综上所述,脓毒症诱导了海马星形胶质细胞向神经毒性的A1表型极化,伴随AQP4表达上调及自噬功能可能受损。
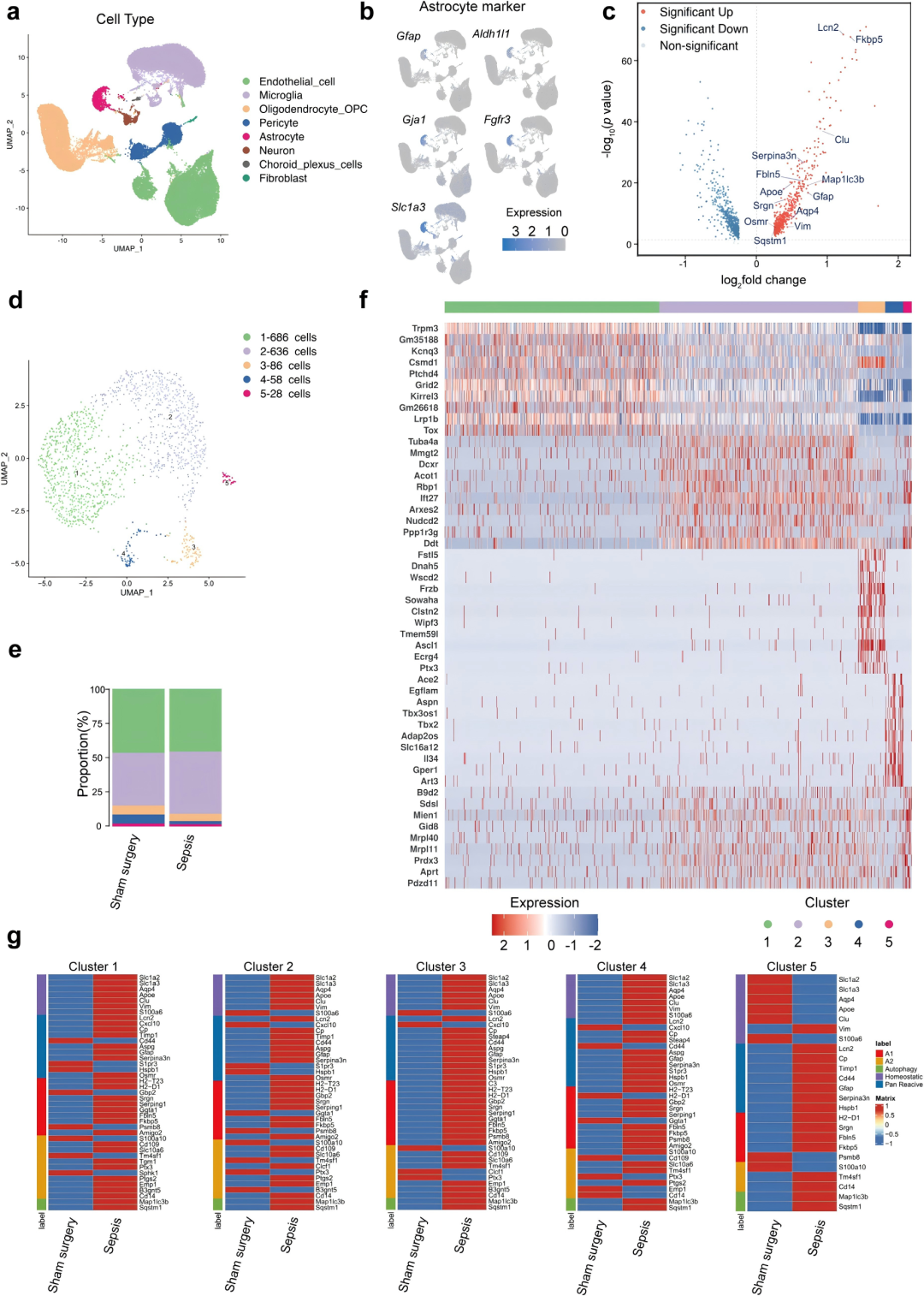
图1. scRNA-seq显示,脓毒症小鼠星形胶质细胞中AQP4及星形胶质细胞反应性上调。
Result 2 脓毒症损害小鼠长期神经认知功能和LC-HP-NA,导致海马星形胶质细胞功能异常
作者通过多层面实验揭示了脓毒症导致长期认知障碍的机制(实验流程见Fig. 2a)。行为学上,脓毒症小鼠在Barnes迷宫测试(训练与测试阶段,Fig. 2b, c)和恐惧条件反射(Fig. 2d)中表现出显著的学习和记忆缺陷。在神经环路层面,脓毒症诱导了蓝斑(LC)去甲肾上腺素能神经元凋亡(TUNEL/TH共标记增加,Fig. 2e),并导致海马区去甲肾上腺素释放及其受体激活信号显著降低(Fig. 2f, g)。在海马组织内,脓毒症引发了显著的病理改变:水通道蛋白4(AQP4)表达上调(Fig. 2h),细胞自噬被抑制(表现为LC3B-II/I比率下降、P62积累及星形胶质细胞中自噬体减少,Fig. 2i, j)。此外,星形胶质细胞反应性增强(补体C3表达增加,Fig. 2k),同时,海马神经元中突触后密度蛋白95(PSD95)和突触素的表达以及树突棘密度均降低(Fig. 2l, m)。上述结果表明,脓毒症通过损害LC-HP-NA系统功能,触发海马星形胶质细胞AQP4上调和自噬抑制,进而增强其反应性并最终损伤突触结构与认知功能。
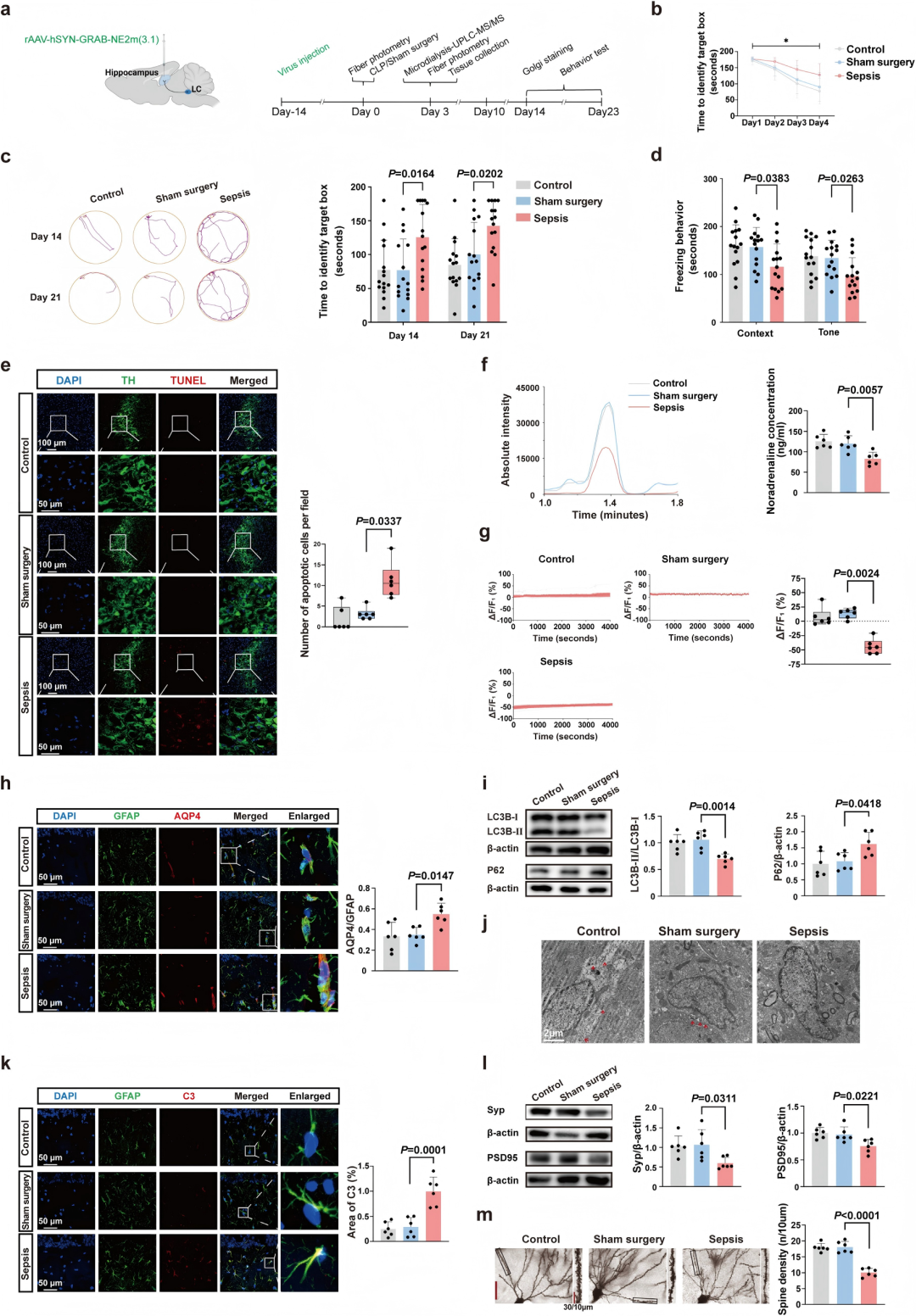
图2. 脓毒症损害了LC-HP-NA,并在海马体中诱导病理学改变,导致长期认知功能障碍。
Result 3 激活LC-HP-NA对脓毒症小鼠产生神经保护作用
作者采用化学遗传学方法特异性激活脓毒症小鼠受损的LC-HP-NA系统(Fig. 3a),探究其治疗潜力。行为学测试表明,该激活干预显著改善了脓毒症小鼠的长期认知功能:在Barnes迷宫中缩短了找到目标箱的时间(Fig. 3b, c),并增强了恐惧条件反射中的僵直行为(Fig. 3d)。机制上,激活成功诱导了蓝斑DBH阳性神经元的c-Fos表达(Fig. 3e),并恢复了脓毒症小鼠海马区的去甲肾上腺素释放水平与受体激活信号(Fig. 3f, g)。在细胞分子层面,激活干预逆转了脓毒症引发的关键病理改变:降低了海马星形胶质细胞中AQP4/GFAP的荧光强度(Fig. 3h),促进了细胞自噬(表现为LC3B-II/I比率升高、P62减少及自噬体增多,Fig. 3i, j),并抑制了星形胶质细胞的反应性(C3表达降低,Fig. 3k)。最终,小鼠海马中PSD95和突触素的表达以及树突棘密度也得以恢复(Fig. 3l, m)。综上所述,化学遗传学激活LC-HP-NA不仅提高了海马去甲肾上腺素水平,减轻了脓毒症诱导的学习记忆损伤,还改善了脓毒症小鼠海马的星形胶质细胞功能。
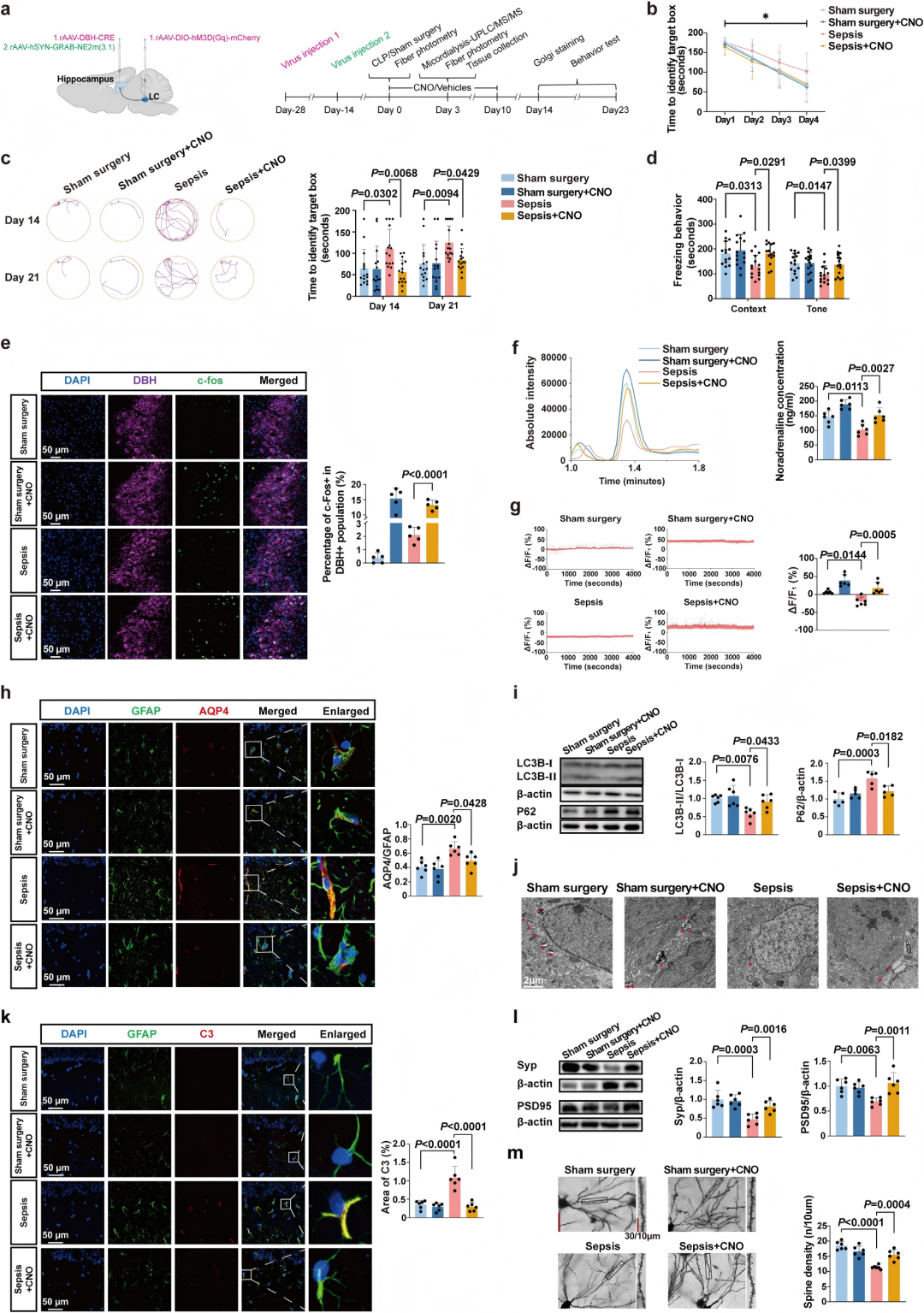
图3. LC-HP-NA的化学遗传学激活改善了脓毒症诱导的长期认知功能障碍及海马病理学改变。
Result 4 激活LC-HP-NA不能对星形胶质细胞α2A-AR敲低的脓毒症小鼠产生神经保护作用
作者研究发现,脓毒症后海马星形胶质细胞中Adra2a+细胞的比例显著上调。为探究LC-HP-NA激活的保护作用是否依赖该受体,作者敲低了海马星形胶质细胞的α2A-AR。结果显示,在α2A-AR敲低小鼠中,激活LC-HP-NA未能改善脓毒症导致的长期认知缺陷,表现为Barnes迷宫和恐惧条件反射测试成绩无提升(Fig. 4a-d)。同时,该保护作用相关的星形胶质细胞功能改善也消失:包括AQP4表达未降低(Fig. 4e)、自噬未增强(LC3B-II/I比率和P62表达无变化,自噬体未增加)(Fig. 4f-g)、星形胶质细胞反应性未减弱(C3阳性区域未减少)(Fig. 4h),以及海马突触结构未恢复(PSD95、突触素表达及树突棘密度无改善)(Fig. 4i-j)。以上结果表明,海马星形胶质细胞α2A-AR介导了LC-HP-NA化学遗传学激活对脓毒症的长期神经认知保护和星形胶质细胞功能改善作用。
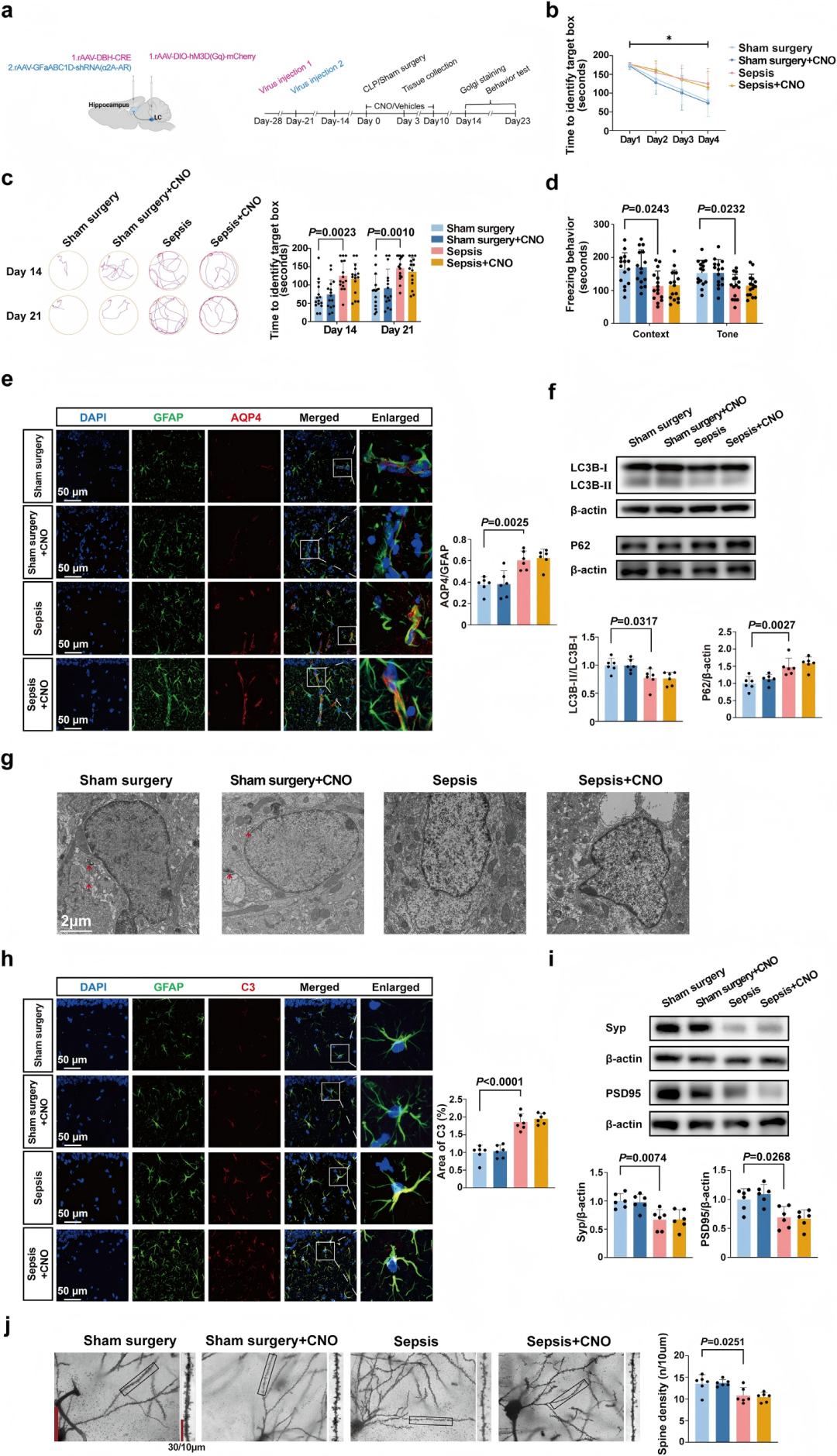
图4. LC-HP-NA的化学遗传学激活未能在星形胶质细胞a2A-AR敲除小鼠中产生长期神经认知保护作用或改善海马病理学改变。
Result 5 激活LC-HP-NA不能对星形胶质细胞AQP4过表达的脓毒症小鼠产生神经保护作用
为验证水通道蛋白4(AQP4)是否为LC-HP-NA激活发挥神经保护作用的关键调节靶点,作者在海马星形胶质细胞中特异性过表达了AQP4。实验发现,在AQP4过表达的脓毒症小鼠中,激活LC-HP-NA未能改善其长期认知功能,表现为Barnes迷宫和恐惧条件反射测试成绩无提升(Fig. 5a-d)。同时,该激活原本对星形胶质细胞功能的改善作用也被阻断:包括自噬未增强(LC3B-II/I比率和P62表达无变化,自噬体未增加)(Fig. 5e-f)、星形胶质细胞反应性未减弱(C3阳性区域未减少)(Fig. 5g),以及海马突触结构未恢复(PSD95、突触素表达及树突棘密度无改善)(Fig. 5h-i)。以上结果证明,海马星形胶质细胞AQP4是介导LC-HP-NA化学遗传学激活对脓毒症的长期神经认知保护和星形胶质细胞功能改善作用的重要分子。
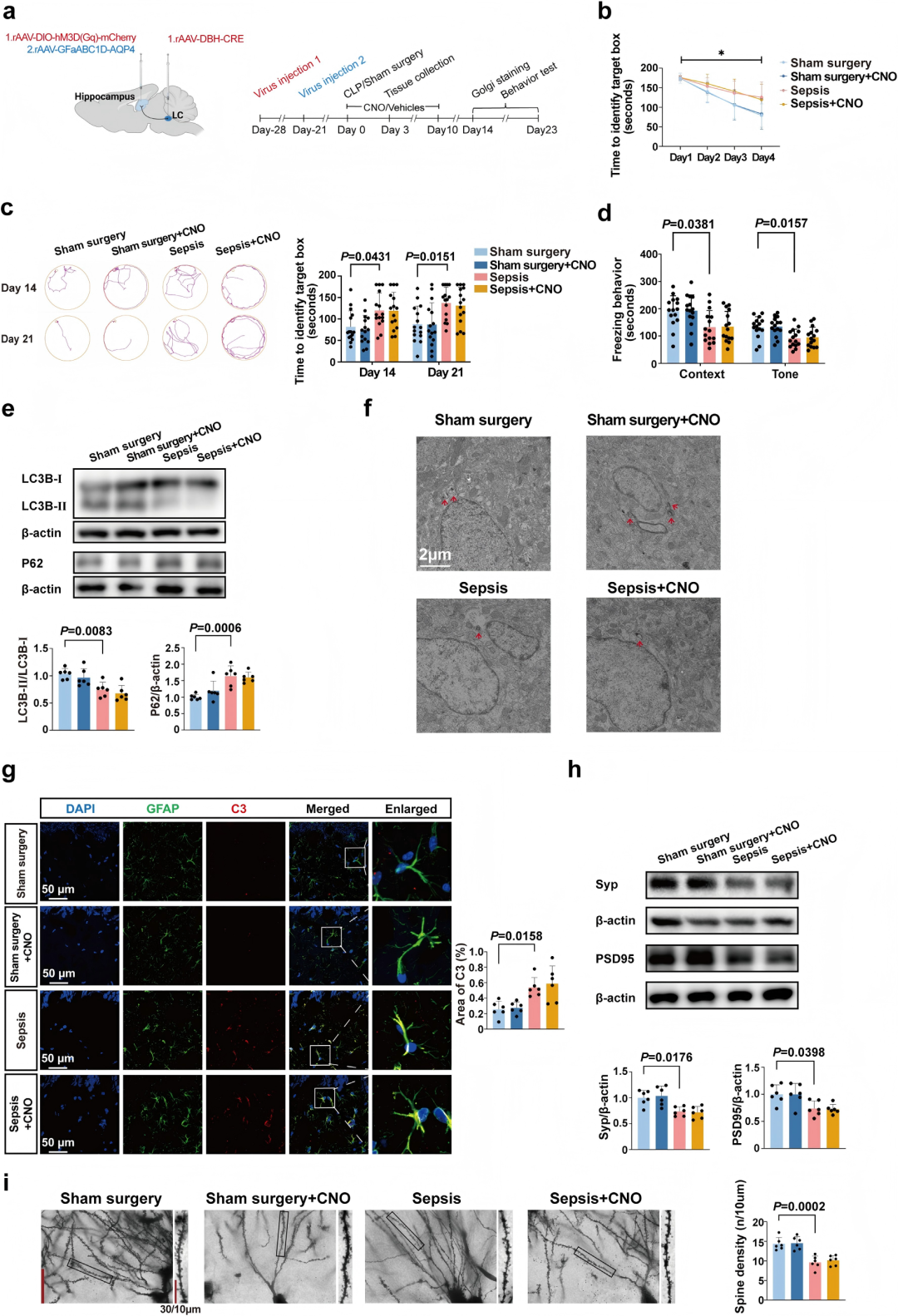
图5. 激活LC-HP-NA的神经保护作用由海马星形胶质细胞AQP4介导。
Result 6 α2A-AR激动剂降低AQP4表达,改善自噬,并抑制LPS刺激的原代星形胶质细胞反应性
作者进一步通过细胞实验表明,α2A-AR激动剂可调控LPS诱导的星形胶质细胞反应性与自噬(Fig. S13a)。LPS刺激后,星形胶质细胞AQP4表达显著升高(Fig. S13b),自噬被抑制(表现为LC3B-II/I比率下降、P62积累及自噬体减少)(Fig. S13b, d),同时核PPAR-γ降低而p-mTOR升高(Fig. S13c),细胞反应性标志C3表达增加(Fig. S13e)。使用α2A-AR激动剂处理能显著逆转上述变化,表明激活该受体可下调AQP4、增强PPAR-γ/mTOR通路相关的自噬,并抑制星形胶质细胞的过度反应。
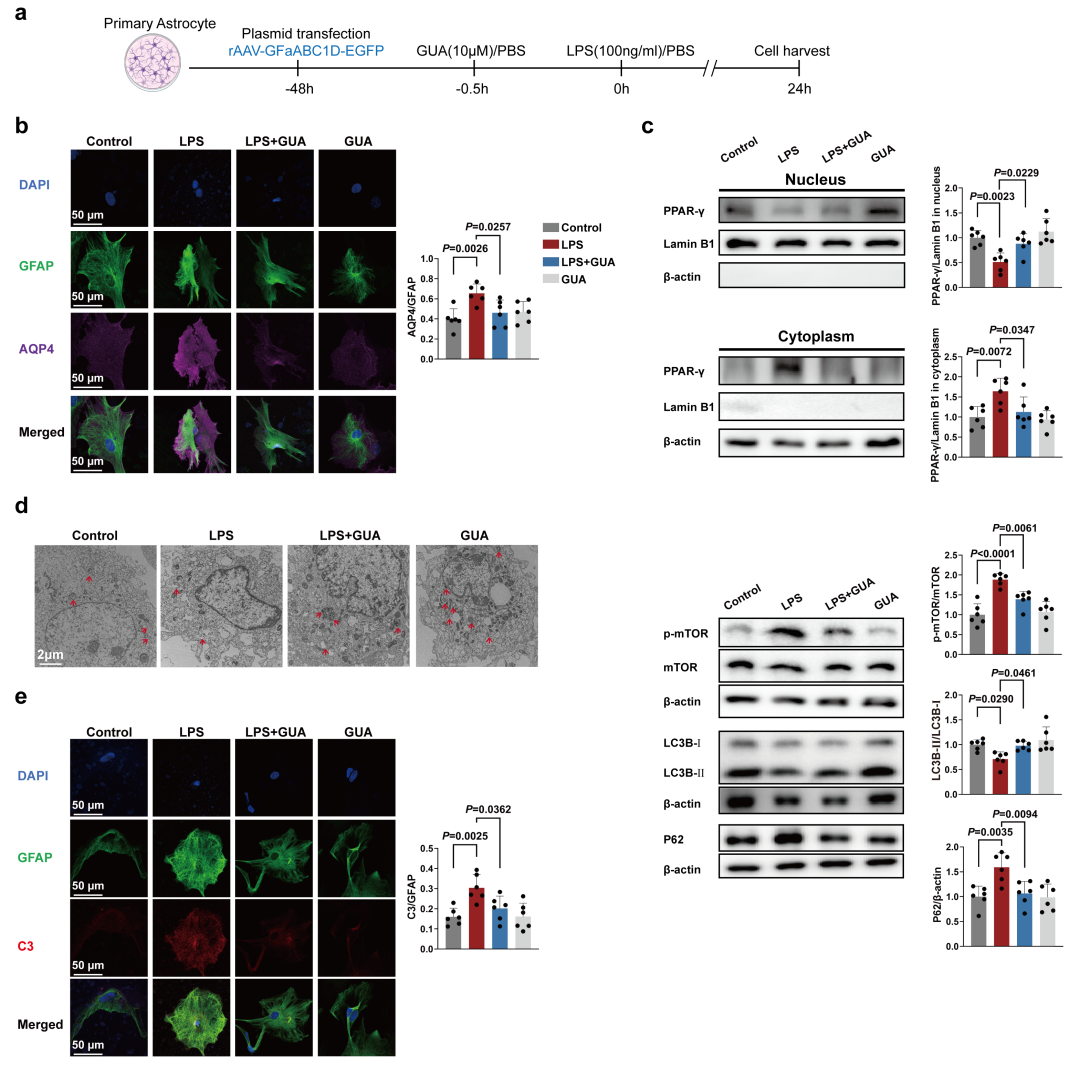
图S13. 激活α2A-AR可减少AQP4的表达,改善自噬,并抑制星形胶质细胞的反应性
Result 7 激活α2A-AR通过AQP4促进原代星形胶质细胞中PPAR-γ/mTOR相关自噬
为验证AQP4是否介导α2A-AR激活对PPAR-γ/mTOR相关自噬的调节,作者在星形胶质细胞中过表达了AQP4。实验发现,在AQP4过表达的细胞中,激活α2A-AR失去了其保护作用:它既不能逆转LPS引起的核PPAR-γ降低和p-mTOR/mTOR比率升高,也无法改善自噬(表现为对LC3B-II/I比率和P62表达的调节作用被减弱)(Fig. S14a-b)。同时,α2A-AR激动剂也未能促进自噬体形成或抑制LPS诱导的星形胶质细胞反应性升高(Fig. S14c-d)。以上结果表明,AQP4介导了α2A-AR激活对PPAR-γ/mTOR相关自噬和星形胶质细胞反应性的调节作用。
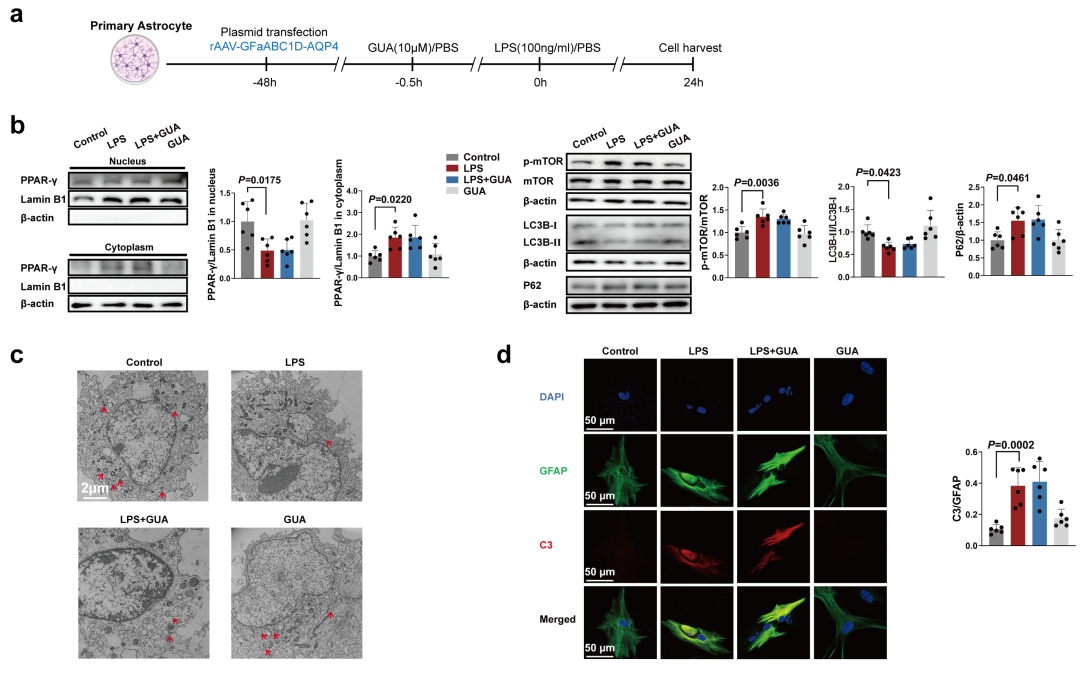
图S14. 激活星形胶质细胞中的α2A-AR可通过AQP4促进星形胶质细胞的自噬。
Result 8 PPAR-γ/mTOR相关自噬调节星形胶质细胞反应性
为确认PPAR-γ/mTOR通路及自噬在调节星形胶质细胞反应性中的作用,作者分别使用PPAR-γ抑制剂GW9662和自噬抑制剂3-MA进行验证(Fig. S15a)。在LPS刺激的星形胶质细胞中,添加GW9662不影响α2A-AR激动剂对AQP4的下调作用(Fig. S15b),但削弱了其对自噬的促进(表现为对LC3B-II/I比率、P62及p-mTOR/mTOR比率的调节作用减弱)(Fig. S15c),并阻断了α2A-AR激动剂抑制细胞反应性的效果(Fig. S15d)。同样,添加3-MA也不影响α2A-AR激动剂对AQP4的调控(Fig. S15e),但减弱了其对自噬指标的改善作用(Fig. S15f),并使其无法抑制LPS诱导的细胞反应性升高(Fig. S15g)。综上,PPAR-γ/mTOR通路介导了α2A-AR对自噬的调节,而自噬的激活是α2A-AR激动剂减轻星形胶质细胞反应性的必要条件。
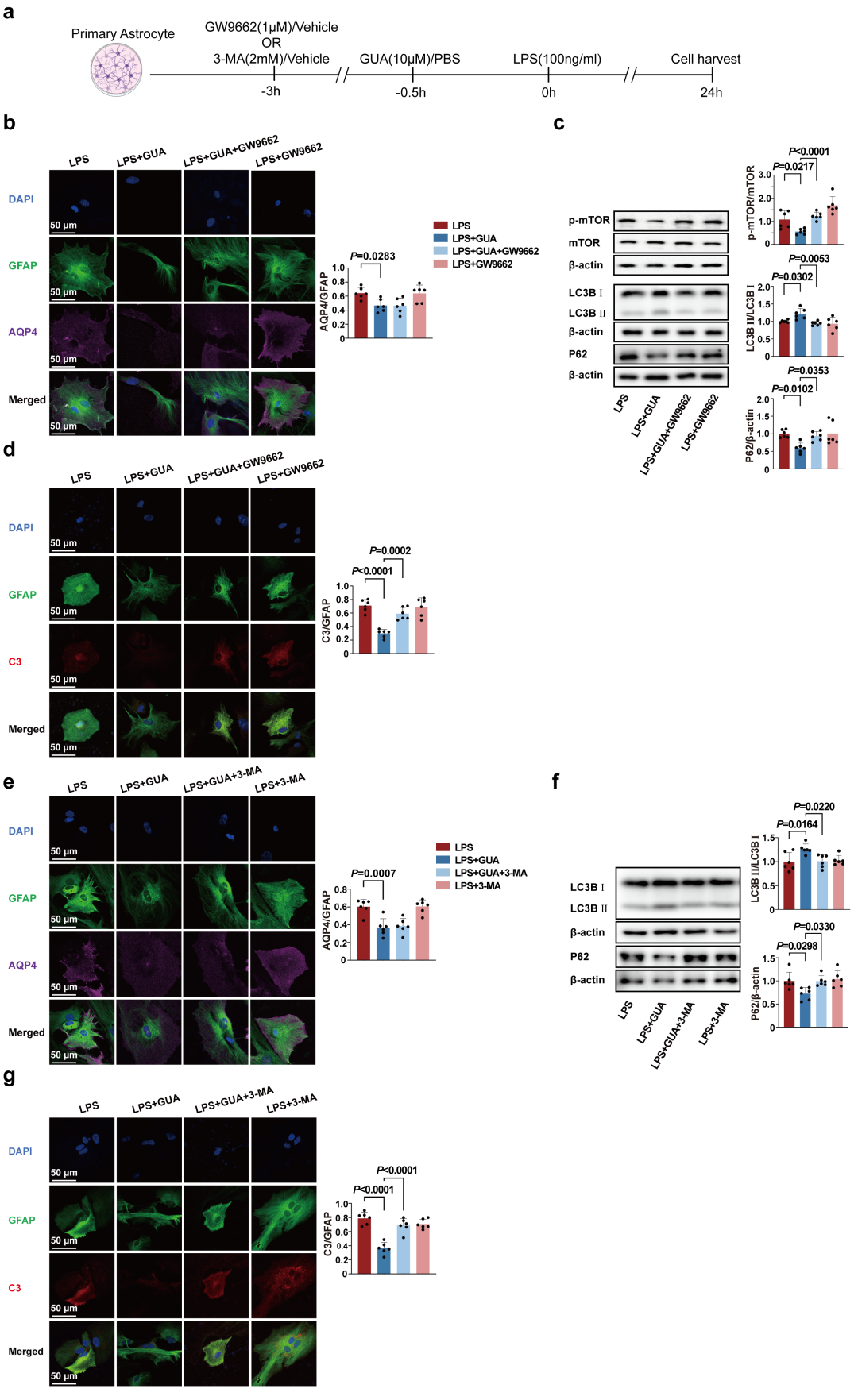
图S15. PPAR-γ/mTOR依赖性自噬调控星形胶质细胞的反应性。
Result 9 激活α2A-AR通过cAMP/PKA信号通路促进星形胶质细胞中AQP4-PPAR-γ/mTOR相关自噬
最后,为探究cAMP/PKA信号通路是否介导α2A-AR激动剂对AQP4表达的调控,作者使用了cAMP类似物8-Br-cAMP(Fig. S16a)。在LPS刺激的星形胶质细胞中,8-Br-cAMP减弱了α2A-AR激动剂对AQP4表达的下调作用(Fig. S16b),并削弱了其对下游信号与自噬的调节,包括对核PPAR-γ、p-mTOR/mTOR比率、LC3B-II/I比率、P62以及PKA Cat表达的影响(Fig. S16c)。此外,8-Br-cAMP还抑制了α2A-AR激动剂促进自噬体形成的能力(Fig. S16d),并使其无法降低LPS诱导的星形胶质细胞反应性标志C3的水平(Fig. S16e)。这些结果表明,cAMP/PKA通路是α2A-AR激动剂调控AQP4表达、进而影响自噬与细胞反应性的关键细胞内信号机制。
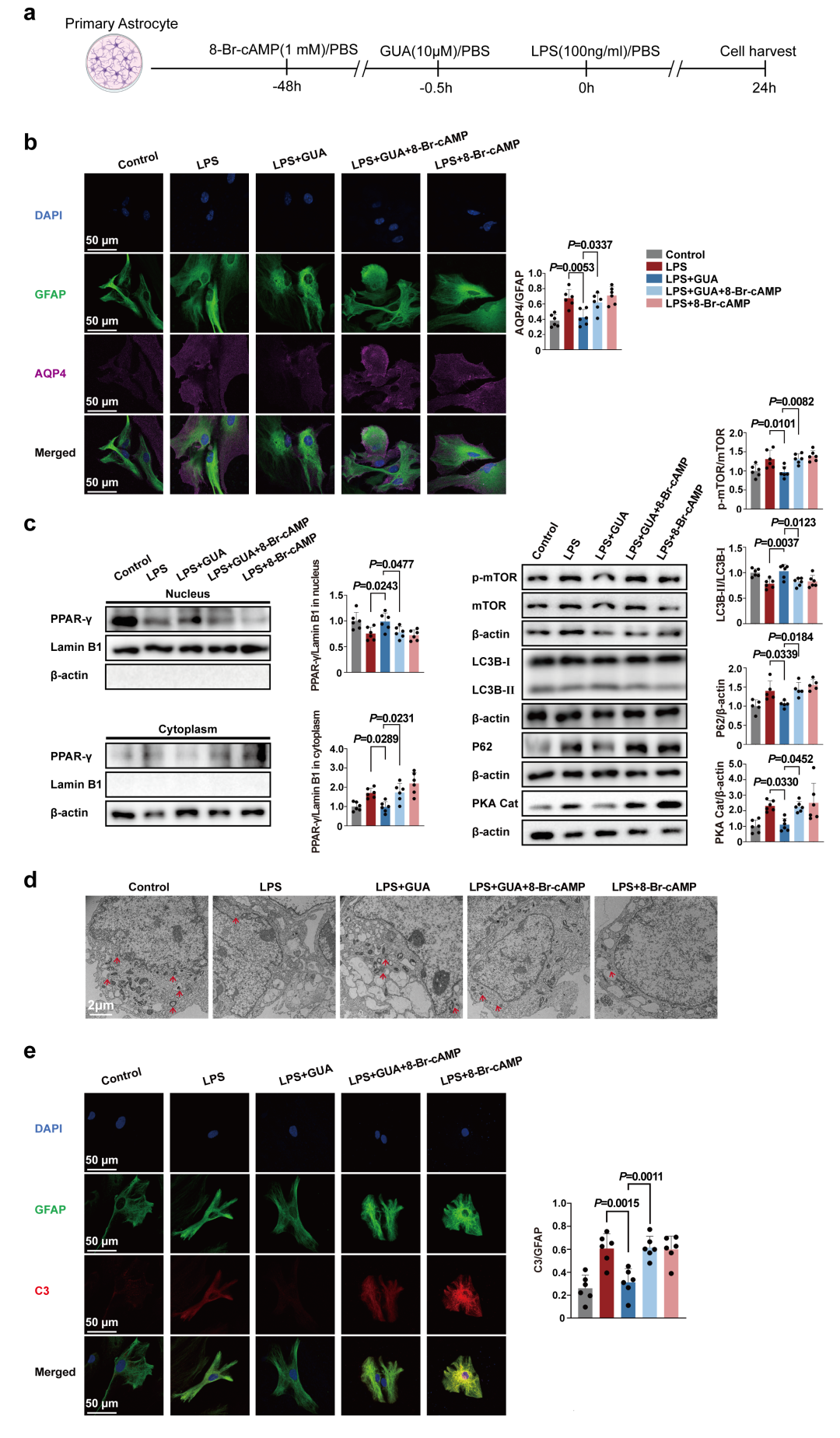
图S16. 通过激活星形胶质细胞中的α2A-AR,经cAMP/PKA信号通路促进PPAR-γ/mTOR依赖性自噬。

研究结论

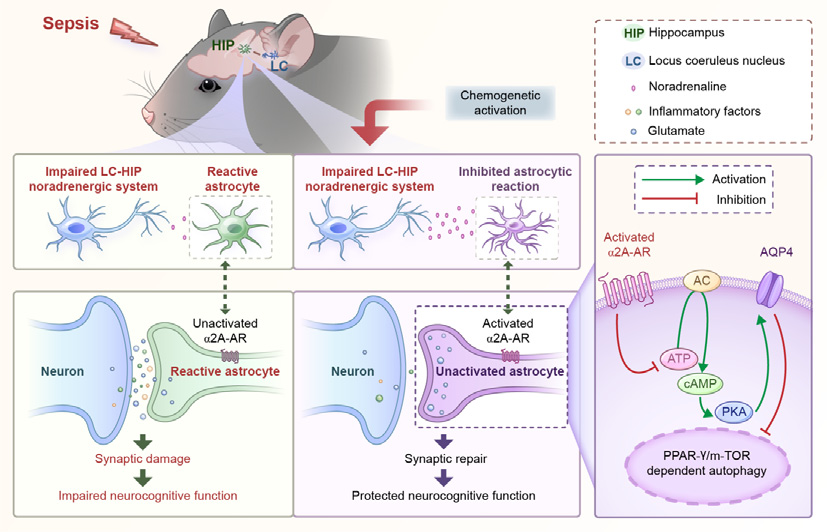
本研究深入解析了LC-HP-NA系统及星形胶质细胞α2A-AR在SAE中的作用与机制。激活LC-HP-NA系统可以通过星形胶质细胞的α2A-AR,经cAMP/PKA信号通路抑制AQP4表达并促进PPAR-γ/mTOR依赖性自噬,从而减轻脓毒症相关脑病中的星形胶质细胞反应性和长期认知功能障碍。为开发针对脓毒症相关脑病的靶向疗法提供了新的理论依据和潜在策略。

参考文献

Juntao Weng, Yueyue Yang, Chenyu Li, Sandong Cao, Xiaoxia Xu, Gaolin Qiu, Dijia Wang, Xiaowen Zheng, Hu Liu, Zhilai Yang, Jiqian Zhang, Qunlin Zhang, Yao Lu, Qiying Shen, Daqing Ma, Xuesheng Liu, Bin Mei. Locus coeruleus-hippocampus noradrenergic activation alleviates sepsis-associated encephalopathy by promoting astrocytic AQP4-related autophagy via α2A-AR. Journal of Advanced Research. 2026, ISSN 2090-1232. https://doi.org/10.1016/j.jare.2026.03.024.