







研究背景

血管钙化是糖尿病患者急性心血管事件不良结局的主要原因。然而,其分子机制尚未完全阐明,也未发现有效的治疗靶点。
支链氨基酸(BCAA)代谢可参与多种心血管疾病,而BCAT2是BCAA分解代谢的限速酶,其在糖尿病动脉钙化中作用及机制不明。
2026年1月6日,江苏大学附属医院王中群教授团队在《Research》(IF=10.7)发表题为 “Vascular smooth muscle cells-specific BCAT2 deficiency attenuates diabetic atherosclerotic calcification via histone propionylation” 的研究论文。该研究整合空间代谢组学、单细胞转录组(scRNA-seq)、Bulk RNA-seq等多组学技术,结合体内外功能实验,系统解析了糖尿病动脉粥样硬化钙化的全新分子调控机制,首次阐明BCAT2介导的支链氨基酸分解代谢在糖尿病动脉斑块钙化中的核心作用及表观遗传调控机制,同时明确了靶向BCAT2在糖尿病动脉斑块钙化治疗中的潜在临床应用价值。
【欧易生物为该研究提供了空间代谢组学技术服务】


研究思路



研究结果

1. 空间代谢组学分析揭示糖尿病足截肢患者钙化胫前动脉中支链氨基酸(BCAA)分解代谢增强
作者首先将糖尿病足截肢患者的胫前动脉分为无血管钙化(NVC)组和血管钙化(VC)组,开展了AFADESI-MSI空间代谢组学分析,发现VC组中BCAA合成与分解代谢通路显著富集。鉴于代谢酶是生物代谢网络的关键节点,也是潜在药物靶点,研究进一步发现,BCAA合成与分解代谢的关键酶BCAT2在VC组中表达变化最为显著,同时该组内缬氨酸、亮氨酸、异亮氨酸这类BCAA水平下降,其代谢产物BCKA(KMV、KIV)水平则相应升高。后续体外实验进一步验证,糖尿病动脉钙化过程中BCAA分解代谢呈增强趋势,且这一过程依赖于细胞对BCAA的摄取增加。
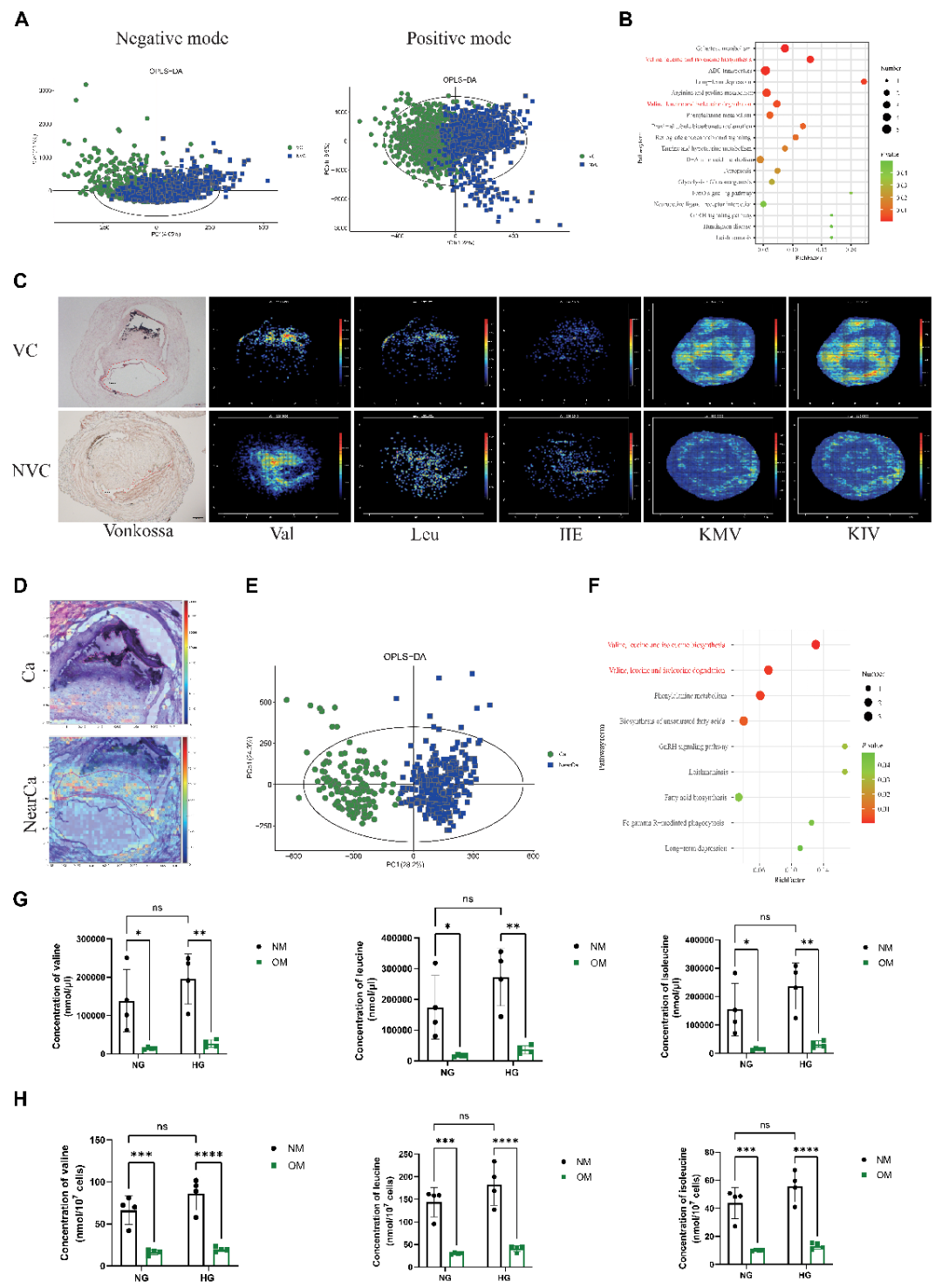
图1. 空间代谢组学揭示BCAA分解代谢增强
2. 糖尿病动脉粥样硬化钙化过程中,BCAT2在血管平滑肌细胞(VSMCs)中高表达
作者采用单细胞测序的方法鉴定出糖尿病截肢动脉组织的9种细胞类型,结果显示BCAT2主要在平滑肌细胞(VSMC)中高表达(VSMC是血管钙化的主要细胞)。作者通过临床样本免疫荧光染色、糖尿病雄性ApoE-/-小鼠体内实验和细胞实验证实了BCAT2在钙化/糖尿病诱导的VSMC中呈现特异性高表达,且BCAT2的mRNA、蛋白水平及酶活性均随血管钙化发生显著上调。
为了探究BCAT2上调的潜在分子机制,研究团队通过结合单细胞调控网络推断分析SCENIC和PROMO工具发现,ATF3可以作为BCAT2的负向转录因子,但在高糖环境下结合效率降低,最终导致BCAT2在VSMC中异常高表达。
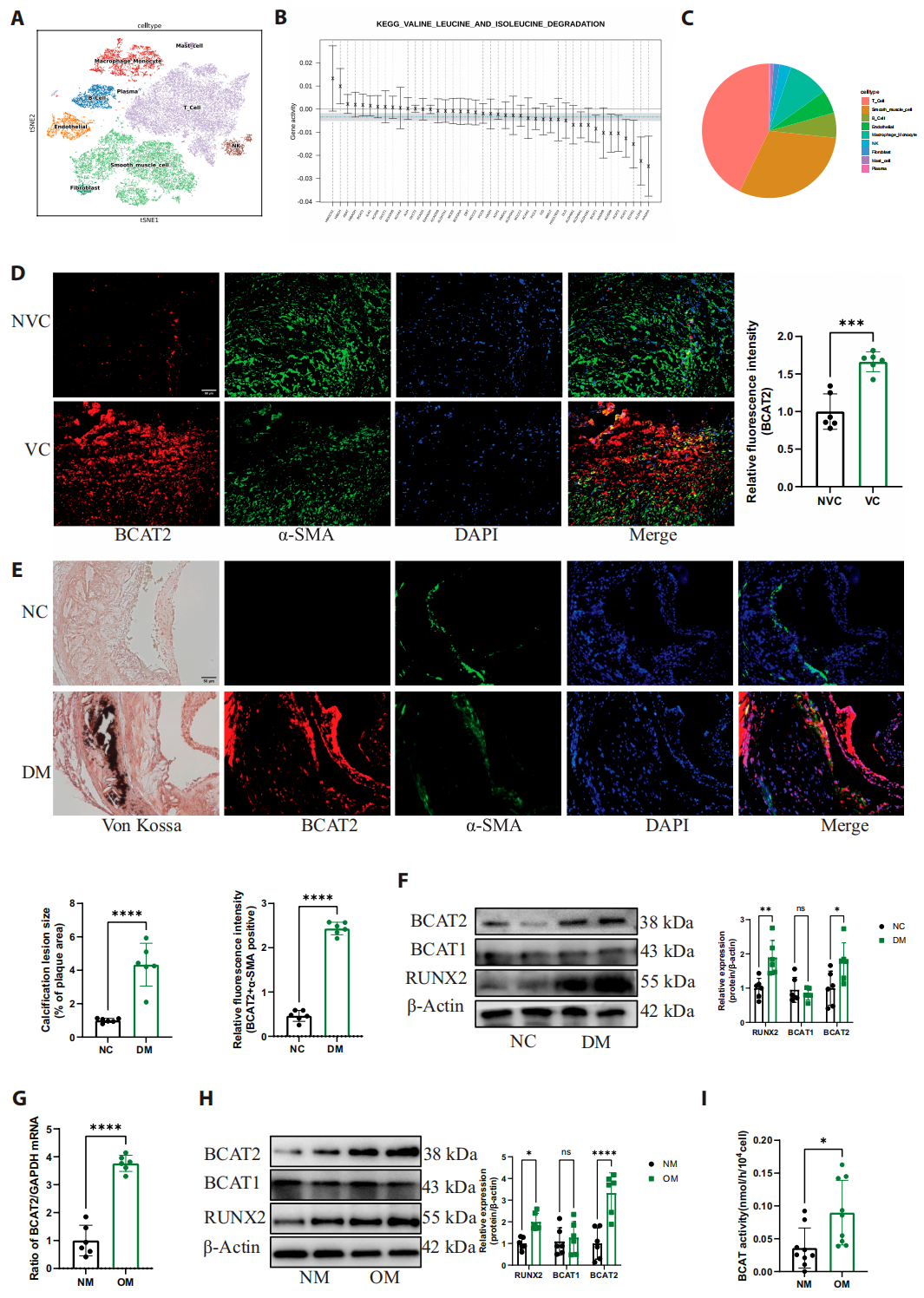
图2.BCAT2在VSMC中高表达
3. 血管平滑肌细胞特异性BCAT2缺失抑制VSMCs成骨分化及糖尿病动脉粥样硬化钙化
为了探究BCAT2在糖尿病斑块钙化形成中的作用,作者构建了ApoE-/-背景下VSMC特异性BCAT2敲除小鼠(ApoE-/-/BCAT2fl/fl/Taglnᶜʳᵉ[ApoE-/-/BCAT2ΔSMC])。研究发现BCAT2敲除后可显著减轻糖尿病诱导的主动脉钙化,降低钙含量和碱性磷酸酶活性,并减少成骨标志物RUNX2和BMP2的表达,同时增加平滑肌标志物α-SMA和SM22α的表达。此外,此效应在雄性和雌性小鼠中均存在,无性别偏倚。
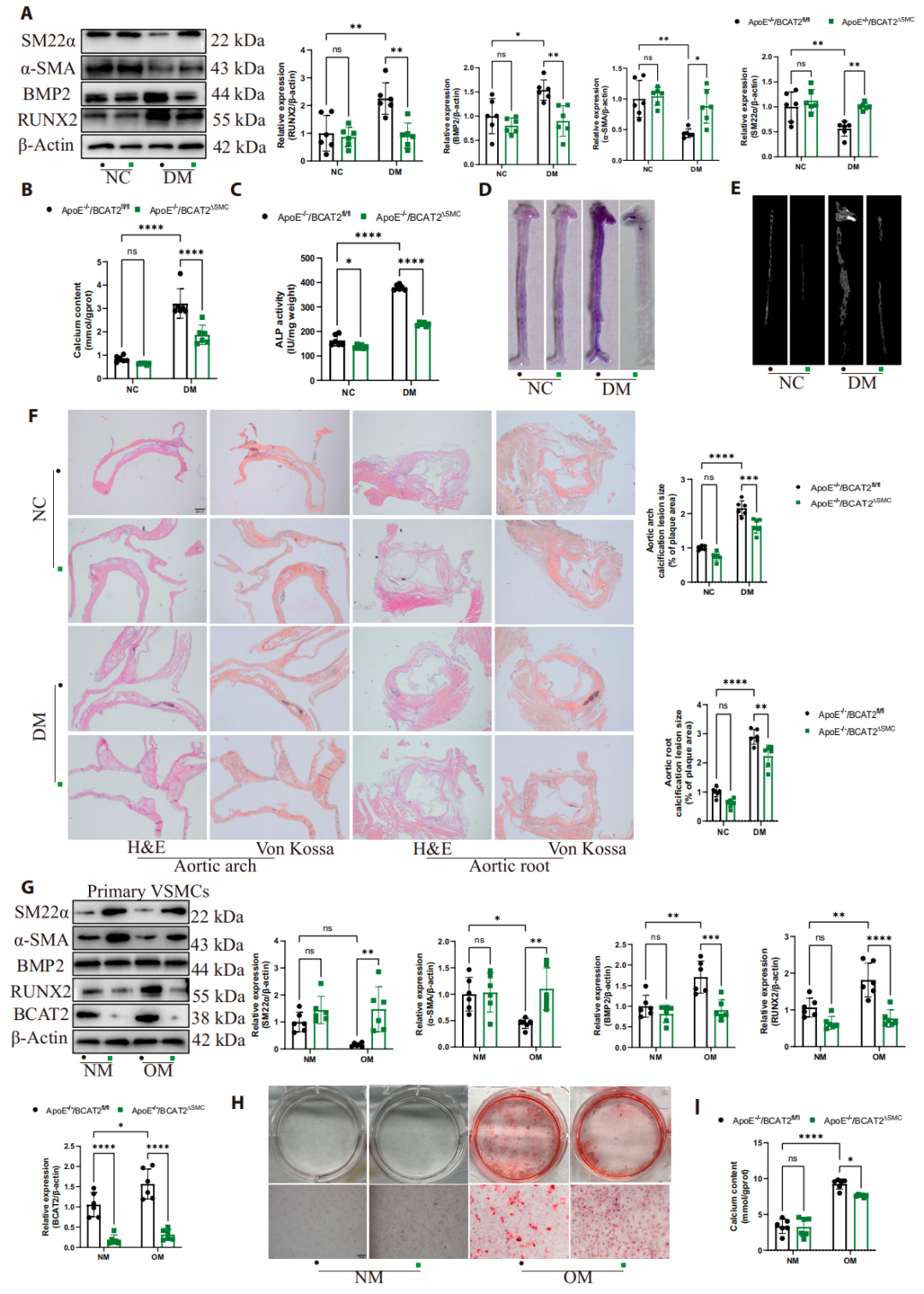
图3.VSMC特异性BCAT2敲除抑制钙化
4. 补充支链α-酮酸(BCKA)可逆转由BCAT2缺失介导的VSMCs成骨分化及糖尿病动脉粥样硬化钙化的抑制效应
为了确定BCAT2介导的支链氨基酸分解代谢在糖尿病动脉粥样硬化钙化中的作用,研究团队给BCAT2敲除小鼠的饮水中补充BCKA,结果发现其可以恢复主动脉的钙化程度以及RUNX2、BMP2的表达。在细胞实验中,BCKA也能恢复VSMC的成骨分化和钙化,证实BCAT2的促钙化作用依赖于其催化生成的BCKA。
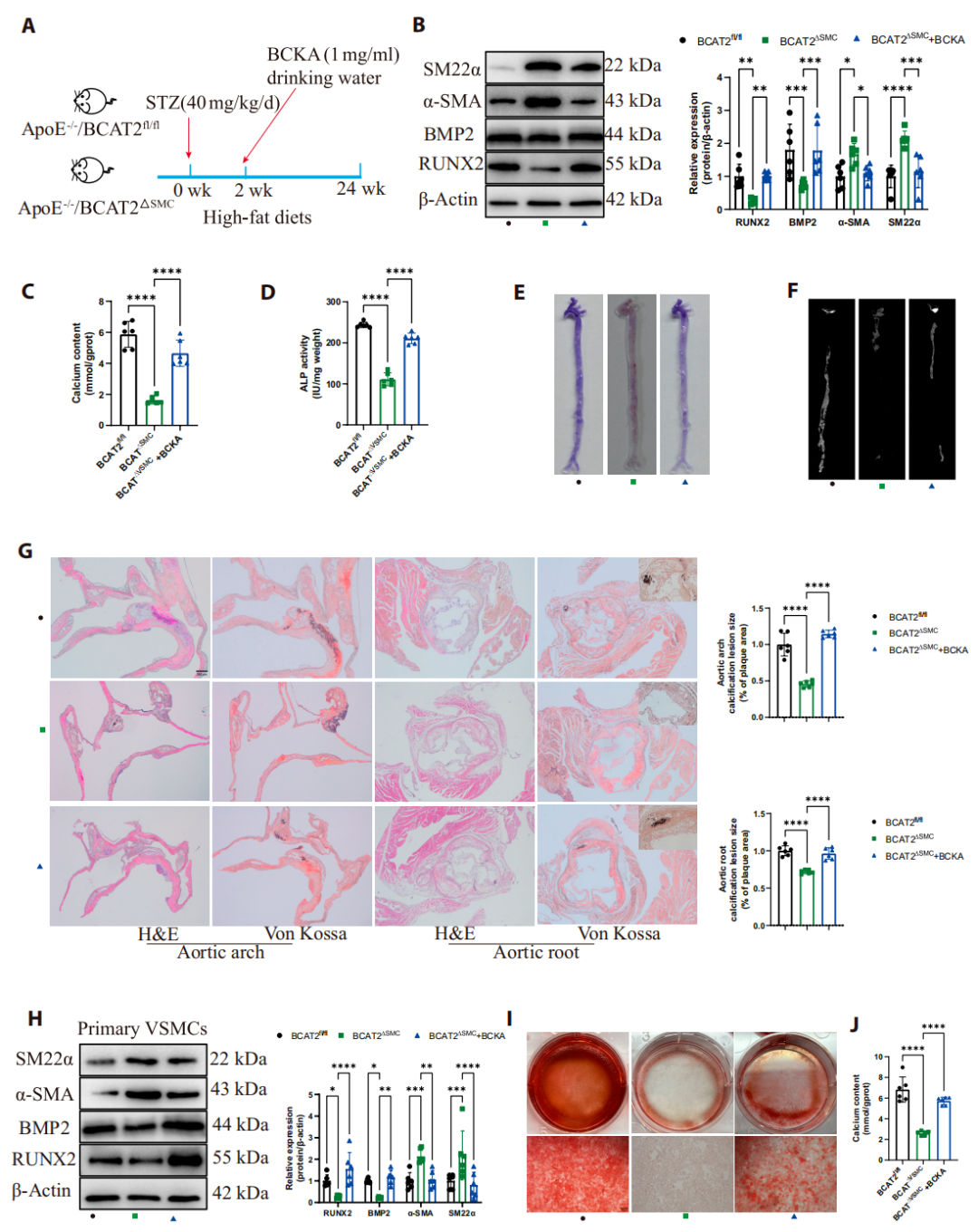
图4. BCKA补充可逆转BCAT2缺失的抑制效应
5. 补充KMV或KIV(而非 KIC)可促进VSMCs成骨分化及糖尿病动脉粥样硬化钙化
研究团队对比三种BCKA的功能差异发现,α-酮-β-甲基戊酸(KMV,异亮氨酸代谢产物)、α-酮异戊酸(KIV,缬氨酸代谢产物)可显著促进血管平滑肌细胞成骨分化及血管钙化进程,而α-酮异己酸(KIC,亮氨酸代谢产物)无明显调控效应。
机制研究表明,KMV与KIV可经支链α-酮酸脱氢酶复合体(BCKD)催化分解生成丙酰辅酶A(prop-CoA),KIC则经该酶催化代谢为乙酰辅酶A;体外外源性补充prop-CoA可模拟KMV、KIV 的促钙化作用,且ApoE-/-/BCAT2ΔSMC小鼠体内prop-CoA 水平显著下调,外源性补充BCKA或prop-CoA可恢复其体内prop-CoA的正常水平;同时,BCAT2敲除对乙酰辅酶A、琥珀酰辅酶A的表达水平无显著影响,上述结果证实prop-CoA是BCAT2-BCKA信号轴介导糖尿病动脉粥样硬化钙化的核心代谢中间体。
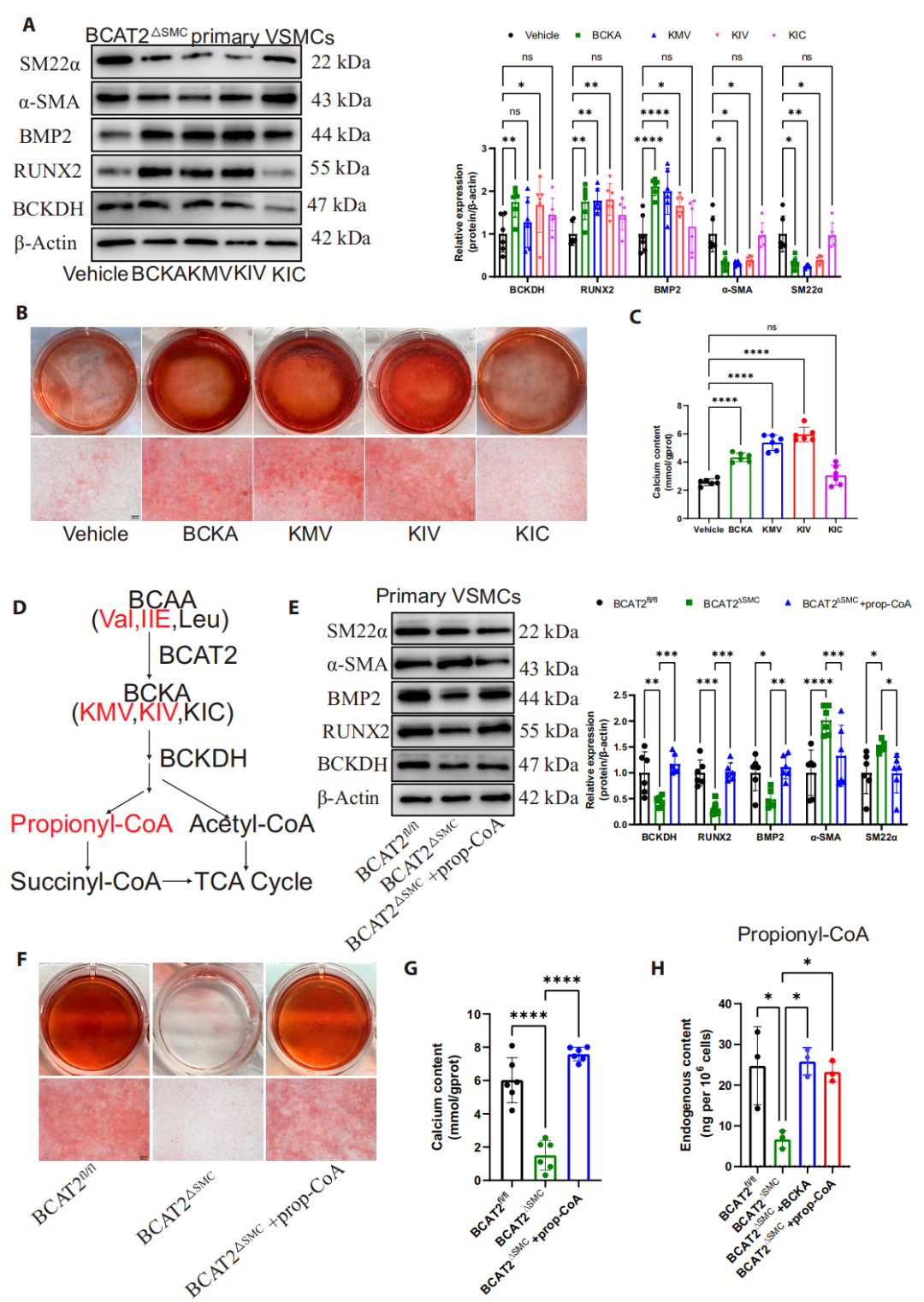
图5.KMV和KIV是主要活性代谢物
6. 血管平滑肌细胞特异性BCAT2缺失抑制组蛋白丙酰化
为了研究prop-CoA调节糖尿病动脉粥样硬化钙化的机制,研究团队检测了VSMC中蛋白质丙酰化水平。结果发现VSMCs特异性BCAT2缺失降低了整体蛋白丙酰化水平,特别是组蛋白H3K23位点的丙酰化(H3K23pr)。而在临床样本中,钙化动脉的 H3K23pr水平显著高于无钙化组。补充prop-CoA可逆转BCAT2敲除介导的H3K23pr降低,证实BCAT2通过调控prop-CoA生成,特异性调节H3K23pr水平。
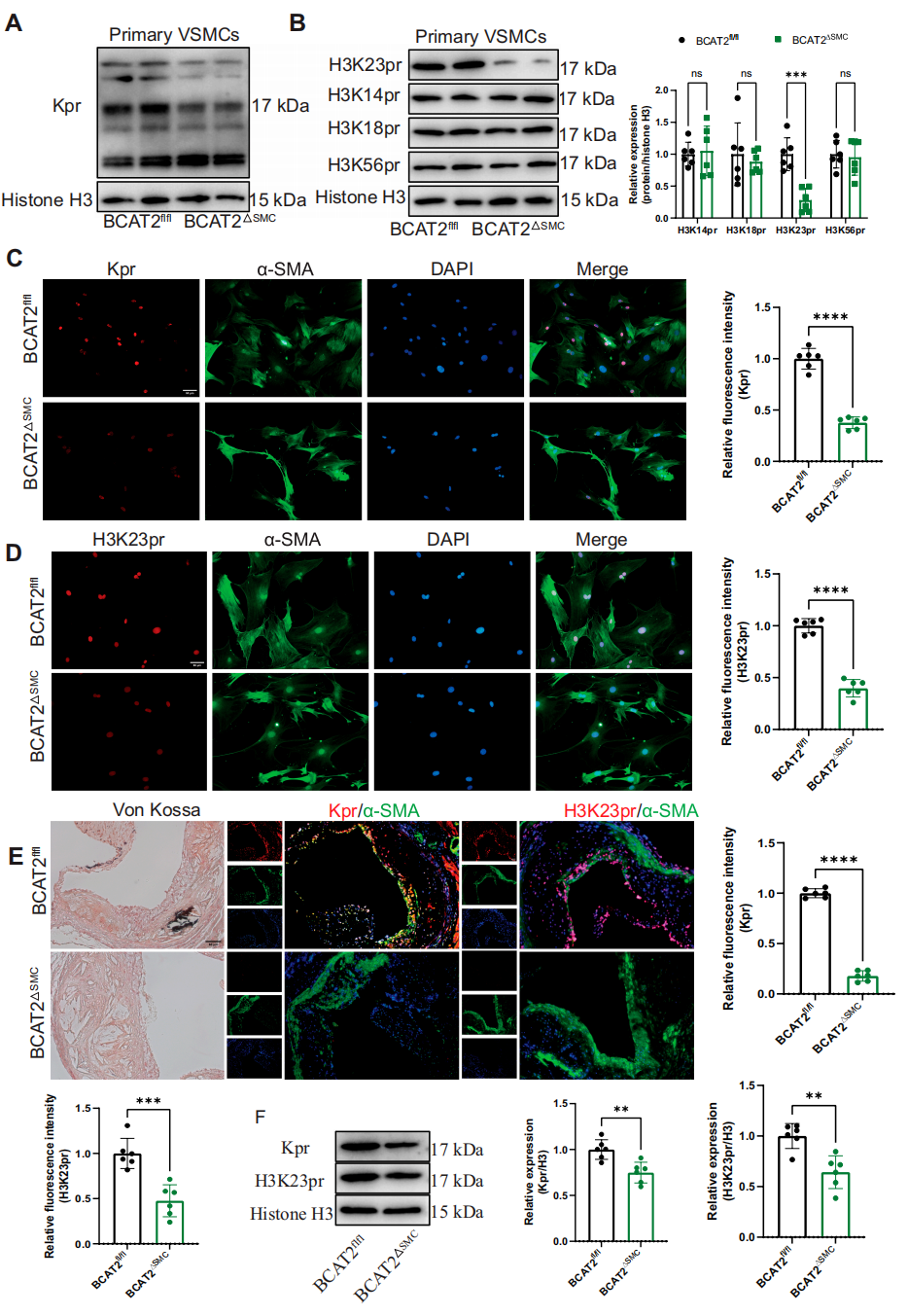
图6. BCAT2缺失抑制组蛋白丙酰化
7. BCAT2-BCKA轴通过组蛋白丙酰化调控RUNX2的表达
接下来作者开展了转录组分析,以表征ApoE-/-/BCAT2ΔSMC小鼠中的转录组变化。结果表明BCAT2敲除小鼠主动脉中包括Runx2在内的多个成骨基因均显著下调。ChIP-qPCR证实H3K23pr富集在RUNX2基因的启动子区,激活RUNX2的转录表达。敲除RUNX2可显著逆转BCKA的促钙化效应:即使补充BCKA,VSMCs的成骨分化标志物仍下调,钙结节形成减少,证实RUNX2是BCAT2-BCKA轴促钙化的下游关键效应分子。
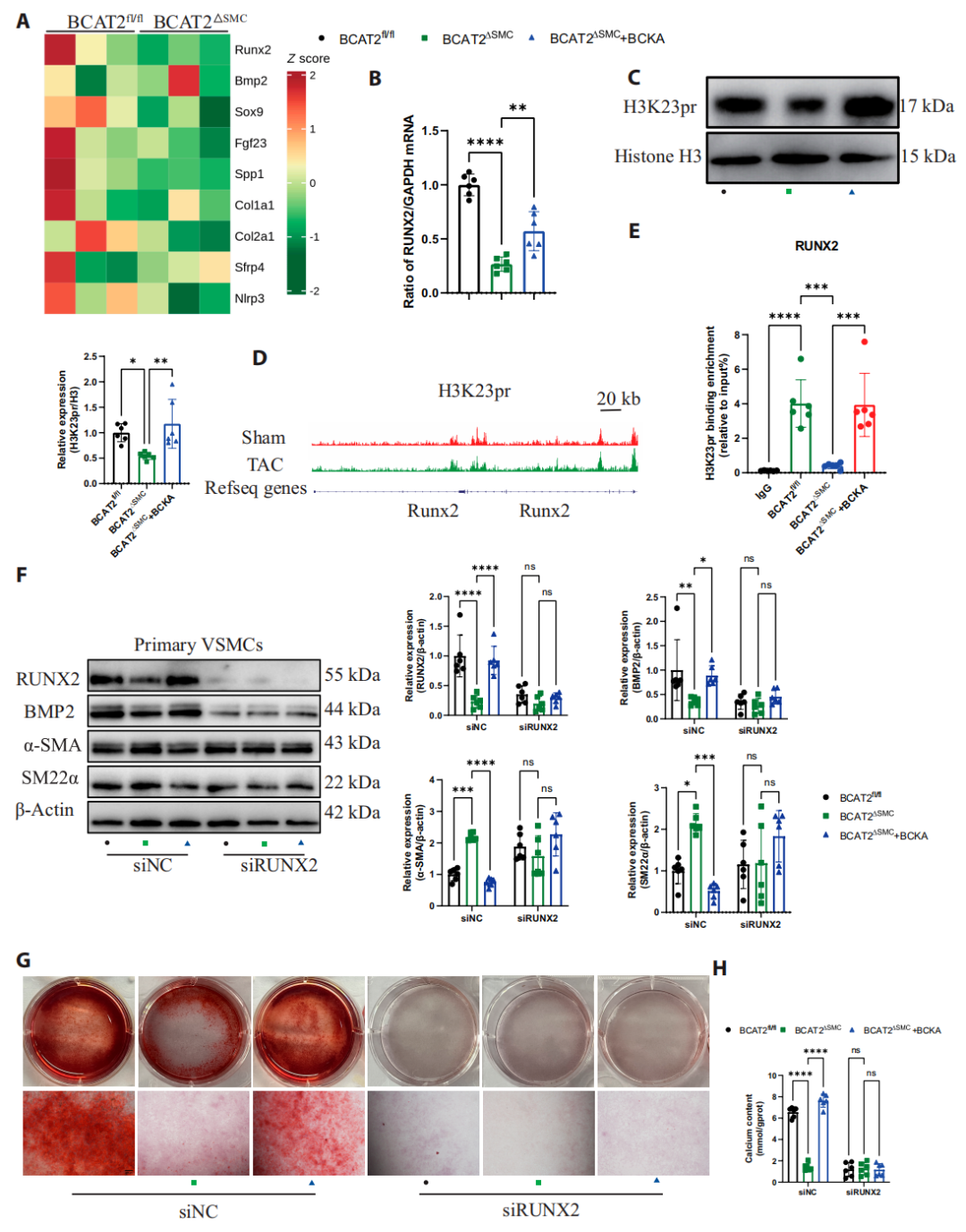
图7.BCAT2-BCKA轴通过组蛋白丙酰化调控RUNX2
8. BCAT2抑制剂可抑制糖尿病动脉粥样硬化钙化
由于BCAT2与糖尿病动脉粥样硬化钙化发展密切相关,研究团队使用BAY-069作为BCAT2的特异性抑制剂,并检测其在治疗糖尿病动脉钙化中的疗效。结果显示体内向糖尿病 ApoE-/-小鼠注射BAY-069,可显著抑制BCAT2的活性和表达,降低糖尿病小鼠主动脉的钙化面积、钙含量和ALP活性,并减少斑块负荷。体外向VSMCs加入BAY-069,亦可显著抑制成骨分化和钙结节形成。结果直接证实靶向抑制BCAT2是治疗糖尿病动脉粥样硬化钙化的有效策略,为临床转化提供了关键实验依据。
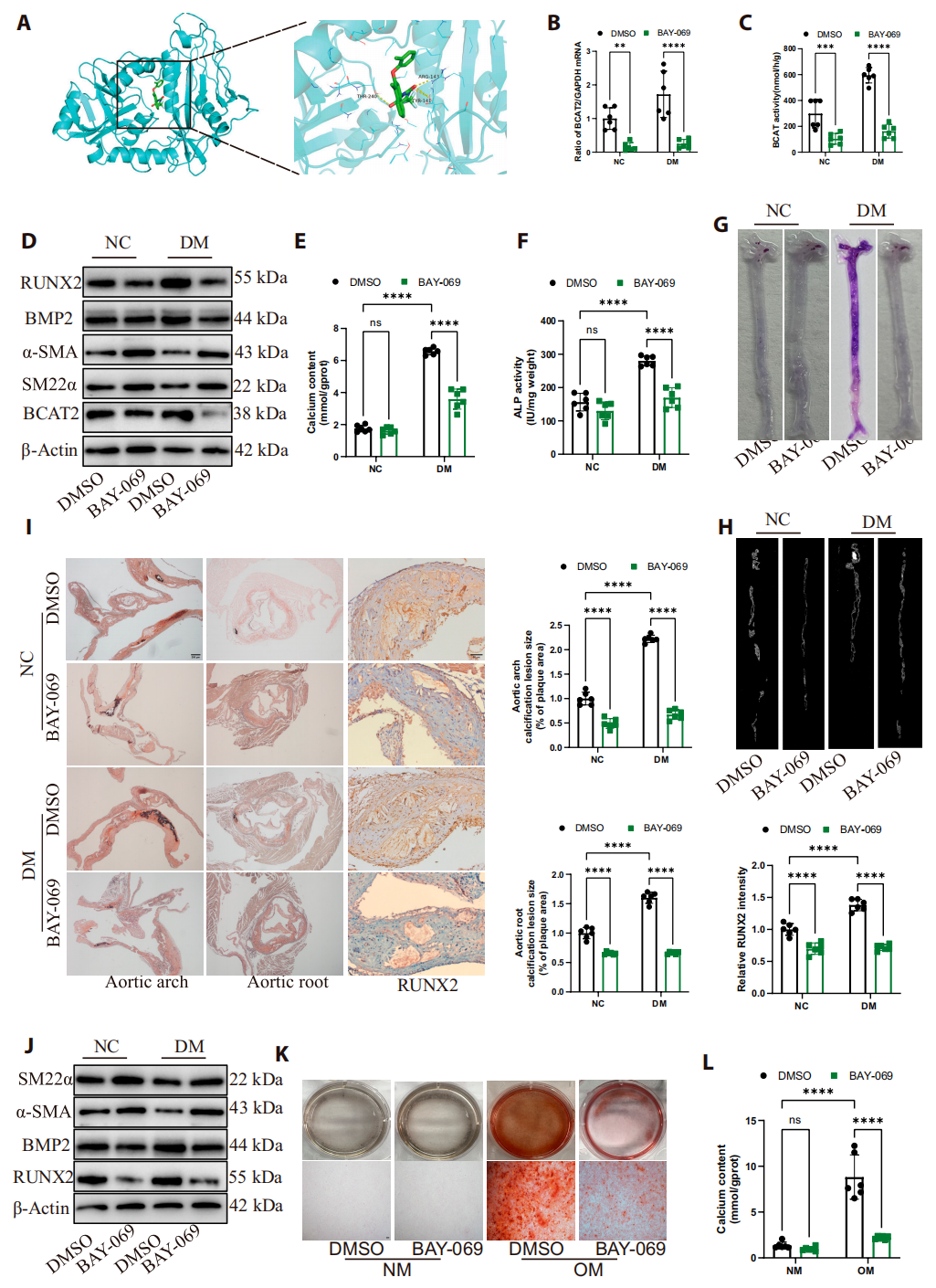
图8.BCAT2抑制剂BAY-069能抑制钙化

研究结论

本研究结合空间代谢组、单细胞转录组、Bulk RNA-seq及体内外实验,解析了糖尿病动脉粥样硬化钙化的全新调控机制,发现糖尿病钙化动脉中BCAA分解代谢增强、VSMC中BCAT2显著上调,证实VSMC特异性BCAT2缺乏可无性别差异减轻糖尿病动脉粥样硬化钙化,而补充KMV、KIV这类BCKA会逆转该保护效应;同时阐明BCAT2-BCKA轴通过调控丙酰辅酶A生成,影响RUNX2启动子区组蛋白丙酰化,进而调控 VSMC成骨分化的核心机制,并验证BCAT2抑制剂BAY-069能有效阻断该通路、显著抑制糖尿病动脉粥样硬化钙化。该研究揭示了BCAA分解代谢在该病中的全新作用,明确BCAT2为潜在治疗靶点,为糖尿病大血管并发症干预提供了新依据。


研究创新点

1.视角创新
首次发现BCAA分解代谢异常激活是糖尿病动脉粥样硬化钙化的重要驱动因素,将支链氨基酸代谢与血管钙化关联,拓展了血管钙化的代谢调控研究范畴。
2.机制创新
首次阐明BCAT2-BCKA-丙酰辅酶A-H3K23pr-RUNX2的代谢-表观遗传调控通路,明确各核心分子节点的特异性作用,填补了该领域机制研究空白。
3.技术与设计创新
融合空间代谢组、单细胞转录组技术,实现代谢物与靶细胞的精准关联;从临床样本到基因修饰动物(无性别偏倚验证)再到细胞实验,结合正反功能验证,形成完整研究闭环。
4.临床转化创新
首次明确BCAT2为糖尿病动脉钙化的潜在治疗靶点,且验证BCAT2抑制剂BAY-069可有效阻断钙化通路,为临床药物研发和疾病干预提供了直接实验依据。